
Neue ICH Q14-Leitlinie wendet QbD auf analytische Methoden an
7 Min. Lesezeit | von Brian Glass
Erschienen im LOGFILE 24/2022
Der neue Leitlinienentwurf (Q14) des International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) für die Entwicklung von analytischen Methoden beschreibt einen erweiterten Ansatz für die Methodenentwicklung, die Validierung und das Lebenszyklusmanagement.
ICH Q2 zur Validierung von analytischen Methoden wurde ebenfalls aktualisiert, um die Änderungen in Q14 widerzuspiegeln. Beide Entwürfe stehen nun zur öffentlichen Konsultation und Kommentierung. Sie stimmen mit dem vorgeschlagenen USP <1220>-Kapitel zum analytischen Lebenszyklusmanagement überein. Neu ist, dass der in Q14 beschriebene erweiterte Ansatz Elemente von Quality by Design (QbD) und Risikomanagement-Tools einsetzt und diese auf analytische Methoden anwendet. Der erweiterte Ansatz ist zwar nicht verpflichtend, aber die Methodik bietet Vorteile.
Traditionelle vs. erweiterte Ansätze zur analytischen Entwicklung
Der herkömmliche Ansatz der analytischen Entwicklung besteht darin, eine Methode zu entwickeln, die funktionieren könnte, und anschließend die Methode zu validieren, ohne alle Faktoren, die die Leistung der Methode beeinflussen, genau zu kennen. Nach der Validierung bleibt die Methode unverändert, bis unweigerlich Probleme auftreten und die Methode geändert und erneut validiert wird.
Die ICH-Leitlinien Q8, Q9 und Q10 beschreiben einen Paradigmenwechsel vom traditionellen Management von Herstellungs- und Qualitätssystemen hin zu einem Ansatz, der sich in erster Linie auf das Verständnis des Prozesses, die Aufrechterhaltung eines Kontrollzustands und die kontinuierliche Verbesserung konzentriert, um die Qualität von Anfang an in das Produkt zu integrieren. Mit dem neuen Leitlinienentwurf wendet ICH Q14 diesen erweiterten Ansatz auf die Entwicklung und Validierung von Analysemethoden an. Dies erfordert einige Anpassungen in der Validierungslandschaft. Der Entwurf der aktualisierten ICH-Leitlinien für die Validierung von Analysemethoden (ICH Q2) spiegelt die Validierungsaktivitäten wider, die zur Umsetzung des erweiterten Ansatzes erforderlich sind.
Der traditionelle Ansatz ist nach wie vor ein gültiges Modell, aber der erweiterte Ansatz hat Vorteile. Aus wissenschaftlicher Sicht besteht der Hauptvorteil darin, dass man einen tieferen Einblick in die kritischen Methodeneigenschaften erhält, was eine größere regulatorische Flexibilität ermöglicht, wenn Änderungen erforderlich sind. Der erweiterte Ansatz ermöglicht die Entwicklung und Validierung eines analytischen Kontrollbereichs, der bei Bedarf die Anpassung einiger Variablen (innerhalb eines Wertebereichs) erlaubt. Ein gründlicheres Verständnis dieser kritischen Parameter führt zu einer robusteren Methodik.
Entwicklung eines analytischen Zielprofils
Der erste Schritt ist die Entwicklung des analytischen Zielprofils (analytical target profile, ATP). Das ATP ist analog zu dem in ICH Q8 beschriebenen Qualitätszielproduktprofil (quality target product profile, QTPP). Dieses Dokument definiert das Ziel der Entwicklung und die gewünschten Leistungsmerkmale der Methode sowie die damit verbundene Unsicherheit. Nach der Entwicklung des ATP verknüpft die Kontrollstrategie kritische Qualitätsattribute (CQAs) mit kritischen Prozessparametern (CPPs). Das ATP dient als Leitfaden für die Entwicklung des Verfahrens und ist der erste Schritt, um sicherzustellen, dass die endgültige Methode für den vorgesehenen Zweck geeignet ist. Wissensmanagement ist ein entscheidender Aspekt des Methodenentwicklungsprozesses – Kenntnisse über die physikalischen und chemischen Eigenschaften des Wirkstoffs und des Arzneimittels sind erforderlich. Zusätzliches Wissen wird auch während des Entwicklungsprozesses erworben und sollte bewahrt werden. Das Risikomanagement wird in allen Phasen des Lebenszyklus des Verfahrens bewertet, um sicherzustellen, dass die Methode unter Kontrolle ist und die ATP-Kriterien erfüllt. Risikobewertungen helfen, die erforderlichen Kontroll- und Replikationsstrategien zu bestimmen.
Robustheit und der erweiterte Ansatz
Bei dem in ICH Q14 diskutierten erweiterten Ansatz geht es um das vollständige Verständnis des Prozesses und der Parameter, die zur Erzeugung der berichtspflichtigen Ergebnisse verwendet werden. Der Schwerpunkt der Robustheitsstudie liegt auf der Entwicklung dieses Verständnisses. Richtig konzipiert, liefert die Robustheitsstudie Informationen darüber, wie sich Anpassungen einzelner Variablen auf das Ergebnis auswirken und wie Variablen mit anderen Variablen interagieren. Beim traditionellen Ansatz wird die Robustheit in der Regel dadurch ermittelt, dass einzelne Faktoren nacheinander variiert werden (one factor at a time, OFAT) und die Auswirkungen der Änderung bewertet werden.
Dieser Ansatz ist zwar immer noch gültig, aber es gehen wichtige Informationen über die Interaktion zwischen Variablen verloren.
Ein chemometrischer Ansatz, der die statistische Versuchsplanung (design of experiments, DoE) und die statistische Analyse nutzt, hat das Potenzial, kritische Variablen zu identifizieren, die sich auf die Methode und die Wechselwirkungen zwischen Variablen auswirken. Die Robustheit wird bei jeder Technik anders umgesetzt, und es wird eine Strategie benötigt, um die Menge der erhaltenen Informationen zu maximieren und gleichzeitig die experimentellen Aktivitäten zu minimieren.
Bei Umkehrphasen-HPLC-Screening-Strategien sind die Variablen auszuwählen, die den größten Einfluss auf den Alpha-Term der allgemeinen Auflösungsgleichung haben, da dieser den größten Einfluss auf die Auflösung hat (vgl. Abb. 1). Faktoren, die sich auf die Selektivität (Alpha) auswirken, sind Säulentyp/Chemie, mobile Phase (pH-Wert, Puffer, Ionenpaarreagenz usw.), organisches Lösungsmittel (MeOH, ACN usw.) und Gradientensteigung oder anfängliche Puffer-/Lösungsmittelkonzentration.
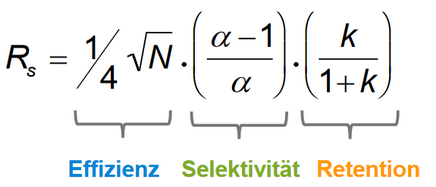
Abbildung 1 Grundgleichung der Auflösung bei der HPLC (Quelle: Agilent Technologies, HPLC – theoretische Grundlagen, 2016) – Abbildung ergänzt durch die Redaktion des GMP-Verlags
Nach dem Screening wird die Methode optimiert, indem die Faktoren, die sich in erster Linie auf den Effizienzterm (N) auswirken, so verändert werden, dass die Bedingungen den Anforderungen der ATP entsprechen. Auf der Grundlage der DoE-Daten wird in einem Vorhersagemodell beschrieben, wie sich Änderungen der Methodenparameter auf die Leistung der Methode auswirken. Statistische Verfahren wie ANOVA (für ein mehrstufiges Regressionsmodell der kleinsten Quadrate) überprüfen die Vorhersagefähigkeit des Modells und liefern Daten zur Rechtfertigung und Validierung des operativen Auslegungsbereichs der Methode (method operational design region, MODR). Anpassungen der Methode innerhalb des MODR bedürfen vor ihrer Umsetzung keiner behördlichen Genehmigung.
Validierung und Lebenszyklusmanagement
Die Revision von ICH Q2 ermöglicht die Anwendung des erweiterten Ansatzes und beschreibt die erforderlichen Validierungsaktivitäten. Die Validierung ist der letzte Schritt im traditionellen Ansatz und wird häufig für jede Methode auf die gleiche Weise durchgeführt und als Formalität behandelt. In begründeten Fällen können Entwicklungsdaten zur Ergänzung des Validierungsberichts verwendet werden, was zu weniger Validierungsaktivitäten führt. Der Ansatz des analytischen Lebenszyklusmanagements konzentriert sich in erster Linie auf die Methodenverzerrung (Richtigkeit) und Präzision der Methode als Teil der Validierung, um die Konformität mit dem ATP zu überprüfen. Während der routinemäßigen Anwendung und Überwachung des Verfahrens werden zusätzliche Daten verfügbar, die zur Bewertung der Leistungsfähigkeit der Methode und der Einhaltung des ATP verwendet werden. Die Überwachung der Leistungsfähigkeit der Methode ist auch für das Lebenszyklusmanagement entscheidend.
Kontinuierliche Herstellung
In den neuen Leitlinien werden auch multivariate Verfahren (MVP) und Echtzeit-Freigabetests (RTRT) behandelt. Dies sind wichtige Aspekte der kontinuierlichen pharmazeutischen Herstellung unter Einbeziehung der prozessanalytischen Technologie (PAT), aber PAT kann auch bei der traditionellen Herstellung (Chargenverarbeitung) eingesetzt werden. PAT-Sensoren messen die physikalischen und chemischen Eigenschaften eines CQAs während des Herstellungsprozesses. Der Input von mehreren Sensoren in Verbindung mit einem multivariaten Modell kann auch CQAs vorhersagen, die nicht direkt messbar sind.
Änderungen nach der Zulassung
Manchmal sind Änderungen an bestehenden Verfahren oder Modellen erforderlich, um die im Laufe der Zeit gewonnenen Erkenntnisse zu berücksichtigen. Diese Änderungen können in jeder Phase des Lebenszyklus auftreten. Beim traditionellen Ansatz werden Änderungen an der Analysemethode den Übewachungsbehörden vor der Umsetzung gemeldet. Je nach Art der Änderung kann die Methode eine vollständige oder teilweise Revalidierung mit anschließender Genehmigung erfordern, bevor die Änderung umgesetzt wird. Ein genehmigtes Änderungsmanagementprotokoll nach der Zulassung (PACMP) und ein Änderungsmanagementplan für den Produktlebenszyklus (PCLM) stellen sicher, dass mögliche Änderungen akzeptiert werden. Dieser Grundsatz spiegelt den Leitfaden ICH Q12 hinsichtlich Änderungen im CMC-Abschnitt von Zulassungsanträgen wider.
Analytische Daten werden in großem Umfang für Zulassungsanträge verwendet, um die Sicherheit und Qualität von pharmazeutischen Produkten nachzuweisen. Da Entscheidungen über die Akzeptanz einer Charge nur so gut sind wie die zur Verfügung gestellten Daten, verringert eine gut entwickelte Analysemethodik, die mit der Kenntnis des Gesamtfehlers bei der Messung einhergeht, das Risiko einer Fehlentscheidung. Mit den neuen Leitlinien verringern die Hersteller das Risiko, eine Charge freizugeben, die nicht den Spezifikationen entspricht.
Der Artikel ist eine deutsche Übersetzung des englischen Originaltextes, der am 13. Mai 2022 von der US-amerikanischen Internetplattform Outsourced Pharma veröffentlicht worden ist: https://www.outsourcedpharma.com/doc/new-ich-q-guidance-applies-qbd-to-analytical-procedures-0001
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de












