

2 Varianten für den Aufbau eines PQR
Auszug aus dem GMP-BERATER, Kapitel 1.J.7.1
8 Min. Lesezeit | von Dr. Christian Gausepohl
Erschienen im LOGFILE 7/2023
„Für die Gliederung des Product Quality Review (PQR) ist es nicht sinnvoll, die 12 Punkte aus dem EU-GMP-Leitfaden in der vorliegenden Form zu übernehmen. Es gilt vielmehr, inhaltliche Blöcke zusammenzustellen, die auch externen Lesern eine einfache Übersicht ermöglichen“, rät Dr. Christian Gausepohl im Wissensportal GMP-BERATER.
Lesen Sie in einem Auszug aus dem GMP-BERATER, welche 2 Varianten unser Autor für den Aufbau eines PQR empfiehlt.
Für die Erstellung des PQR besteht zum einen die Möglichkeit, Produktgruppen zu bilden (z. B. feste Formen bei vergleichbaren Produkten, Wirkstoffgruppen oder verschiedene Dosierungen gleicher Wirkstoffe).
Zum anderen können übergreifende Informationen referenziert werden. Dies erfolgt üblicherweise für nicht produktspezifische Aspekte, z. B. für den jeweiligen Qualifizierungsstatus von Mediensystemen wie Druckluft oder Wasser oder von Multipurpose-Anlagen. Wenn solche Verweise auf andere Systemreviews (z. B. Monitoringberichte) gemacht werden, muss sichergestellt sein, dass diese zum Zeitpunkt der Erstellung des PQR auch tatsächlich vorliegen. Hierzu kann dann auf die spezifischen Dokumentennummern verwiesen werden. Produktrelevante Abweichungen und Auffälligkeiten werden im PQR aufgeführt.
Um die Lesbarkeit des Berichtes zu erhöhen, kann es sinnvoll sein, die Einzeldaten und statistischen Kennzahlen in einem Anhang zu sammeln und ausschließlich die Ergebnisbewertungen im Hauptteil (PQR) zu präsentieren. Zudem kann durch den Fokus auf die Bewertungen im Hauptteil der Aufwand durch den Übertrag von Daten in den Bericht reduziert werden.

Die Gliederung kann sich zudem an den unternehmensspezifischen Prozessen orientieren.
Beispiel 1: Modularer Aufbau
Im nachfolgenden Beispiel (Abbildung 1) wird grob unterschieden zwischen Teilen, die primär durch die Qualitätssicherung (Quality Review), die Herstellung (Technical Review) und die Qualitätskontrolle (Analytical Review) bearbeitet werden bzw. für die diese inhaltlich verantwortlich sind.
Innerhalb eines Teiles werden die Ergebnisse des Abschnitts jeweils am Ende des Kapitels zusammengefasst. Eine Gesamtzusammenfassung findet sich im ersten Teil (Executive Review). Dies erhöht die Lesbarkeit des Dokuments und verbessert die Orientierung. In den Kapiteln selbst finden sich ausschließlich bewertende Kennzahlen. Die entsprechenden Einzeldaten (z. B. Ausbeuten individueller Chargen, analytische Daten) sind zusammen im Anhang abgelegt.
Diese Art von Gliederung bietet sich vor allem dann an, wenn die Herstellung bzw. Prüfung durch einen Auftragnehmer ganz oder teilweise erfolgt und Inhalte so von den Partnern zusammengefügt werden.

Abbildung 1 Beispiel: Modularer Aufbau eines PQR
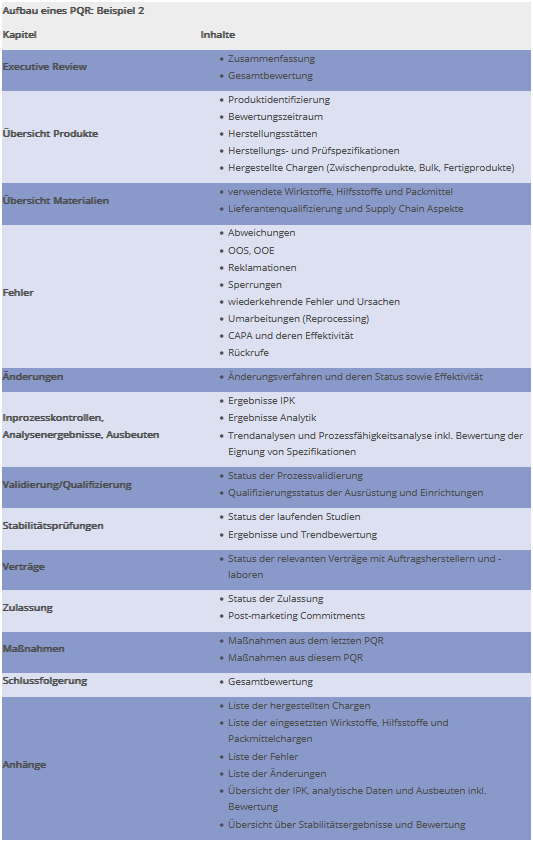
Beispiel 2: Thematischer Aufbau
Eine andere Möglichkeit bietet sich bei einer eher zentralen Erstellung der Bewertung, z. B. durch die Qualitätssicherung unter Verwendung von Einzelberichten aus den Fachbereichen. Hier kann die Lesbarkeit des Berichts im Vordergrund stehen und die Gliederung rein thematisch erfolgen. Das Beispiel in Abbildung 2 zeigt eine mögliche Gliederung.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











