
Die Kontaminationskontrollstrategie (CCS) – mehr als nur ein erweiterter Site Master File
Bericht vom GMP-PharmaCongress 2026
4 Min. Lesezeit | von Dr. Sabine Paris
Erschienen im LOGFILE 08/2026
Eine Kontaminationskontrollstrategie (CCS), wie sie in Anhang 1 des EU-GMP-Leitfadens gefordert wird, sollte einen umfassenden, zugleich aber prägnanten Überblick über die kritischen Kontrollpunkte geben. Sie ist nicht als erweiterte Version des Site Master File gedacht. Vielmehr muss sie wissenschaftlich fundiert, risikobasiert und klar strukturiert sein.
Der GMP-PharmaCongress 2026 fand vom 24. bis 25. März 2026 in Wiesbaden statt. Die Veranstaltung bot acht parallele Konferenzreihen zu hochaktuellen GMP-Themen, darunter aseptische Herstellung, Barrieresysteme, Digitalisierung und künstliche Intelligenz sowie Arzneimittel für neuartige Therapien (ATMPs). GMP-Inspektor Dr. Frank Sielaff (Hessisches Landesamt für Gesundheit und Pflege in Darmstadt) teilte zum Thema Kontaminationskontrollstrategie (CCS) praktische Erkenntnisse aus Inspektionen und stellte Mängel bei der Implementierung der CCS vor.
In der Praxis festgestellte Mängel
In den Inspektionen wurden mehrere typische Schwachstellen identifiziert:
Unzureichende Risikobewertungen
Risikobewertungen waren so konzipiert, dass Risiken von vornherein ausgeschlossen wurden. So führte beispielsweise die Vergabe einheitlich niedriger Schweregrade ohne wissenschaftliche Begründung zu irreführenden Ergebnissen. Darüber hinaus fehlten bei als FMEA bezeichneten Methoden oft wesentliche Elemente der FMEA wie definierte Fehlermodi oder die Neuberechnung von Risikoprioritätszahlen (RPNs) nach der Umsetzung von Abhilfemaßnahmen.
Mängel bei aseptischen Prozessanlagen
In einem Fall wies eine RABS-Abfülllinie mehrere Schwachstellen auf:
- Offene Eingriffe mit unzureichendem Schutz kritischer Komponenten
- Unangemessene Arbeitspraktiken des Bedienpersonals (z. B. Kontakt mit kritischen Oberflächen)
- Unzureichende Einbindung der Qualitätssicherung in aseptische Prozesssimulationen
- Lücken in der Qualifikation des Personals und den Probenahmestrategien
- Unzureichende Konzepte für die Desinfektion und das Umgebungsüberwachung
Unvollständiger Inhalt der CCS
- In einem weiteren Beispiel fehlten wichtige Elemente in der CCS eines aseptischen Isolatorprozesses, darunter:
- Sollwerte, Kritikalität und Alarmverzögerungen für Druckunterschiede
- Details zur Integritätsprüfung der Isolatorhandschuhe
- Einstellwerte für die Luftgeschwindigkeit im Isolator
- Sterilisation von indirekten Kontaktteilen
- Transfer von indirekten Kontaktteilen
- Montage von Maschinenteilen (nur mit Handschuhen im geschlossenen Isolator?)
- Ergebnis der Risikobewertung
- PUPSIT wurde nicht durchgeführt (aufgrund einer möglichen Kontamination durch die PUPSIT-Durchführung; die Risikobewertung befasste sich jedoch nicht mit den Risiken, die sich aus der Nichtdurchführung von PUPSIT ergeben)
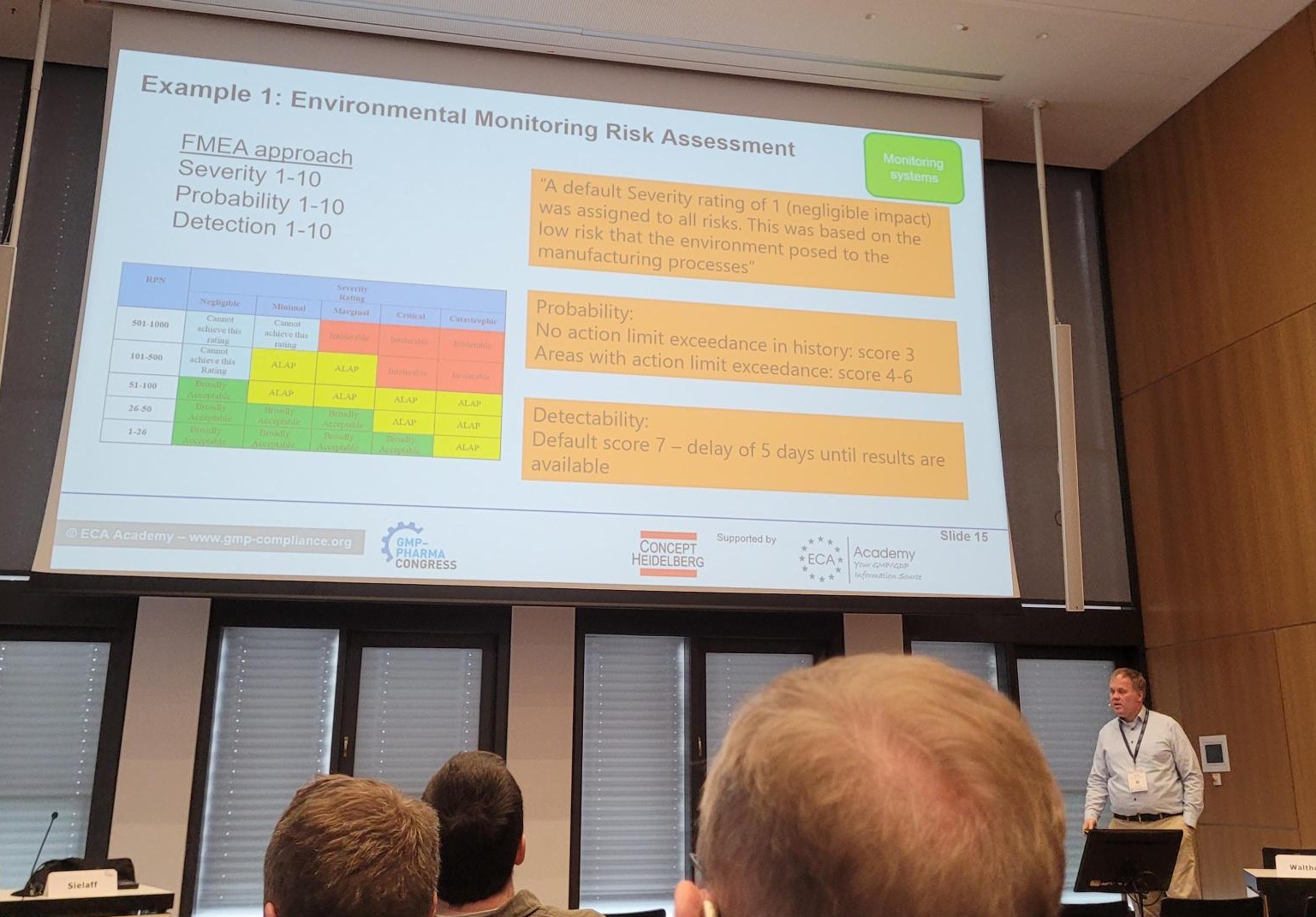
Abbildung 1 | Dr. Frank Sielaff zeigte Beispiele für eine mangelhafte Umsetzung der CCS.
Was Inspektoren erwarten
Frank Sielaff fasste seine wichtigsten Erwartungen an ein wirksames CCS zusammen:
Was zu vermeiden ist
- Allgemeine, unkonkrete Beschreibungen der Aktivitäten am Standort
- Ungefilterte Informationen von Anlagenherstellern ohne Kontext
- Übermäßig detaillierte, irrelevante Informationen
Was wichtig ist
- Prägnante Beschreibungen kritischer Aspekte
- Klare Verweise auf Risikobewertungen, Validierungen und SOPs
- Übereinstimmung mit den Erwartungen von Anhang 1
- Festgelegte Verfahren für die Erstellung und Überprüfung des CCS
Das CCS in Kürze
- Wissenschaftlich fundierte und aussagekräftige Risikobewertungen
- Klare Struktur und logischer Ablauf
- Transparente Verweise auf Begleitdokumente
- Festgelegte Review-Verfahren und deren Auslöser
- Eine solide Grundlage für Inspektionen und Audits – bildet zusammen mit dem Site Master File den ersten Eindruck eines Unternehmens
Fazit
Die CCS ist kein beschreibendes oder gar ein Marketingdokument. Es handelt sich um eine zentrale, risikobasierte Kontrollstrategie, die ein umfassendes Verständnis der Kon-taminationsrisiken und deren Beherrschung nachweisen muss. Ein prägnantes, gut strukturiertes und wissenschaftlich fundiertes CCS ist für die GMP-Compliance und die Inspektionsbereitschaft unerlässlich.
Haben Sie Fragen oder Anregungen? Bitte kontaktieren Sie uns.redaktion@gmp-verlag.de











