

Fehlerbewertungslisten und Fehlerbildkataloge selbst erstellen – aber wie?
Fehlerbewertungslisten und Fehlerbildkataloge
7 Min. Lesezeit | von Dr. Felix Tobias Kern und Fritz Röder
Erschienen im LOGFILE Leitartikel 34/2021
Fehlerbewertungslisten und Fehlerbildkataloge sind beliebte GMP-Dokumente, die seit mehr als 45 Jahren Anwendung finden.
Sie dienen als Rationale für die Qualitätsbeurteilung individueller Chargen und setzen Qualitätsstandards für die Herstellung und für die Testung von pharmazeutischen Produkten. Dabei stellen Sie Life-Cycle Dokumente dar, die mit dem Prozess leben und entsprechend aufgetretener Fehlerbilder auf dem aktuellen Stand gehalten werden müssen.
Für pharmazeutische Herstellbetriebe besteht die Möglichkeit, standardisierte kommerzielle Fehlerbewertungslisten und Fehlerbildkataloge zu kaufen. Diese besitzen allerdings den Nachteil, dass sie auf den vorliegenden Herstellprozess, das Produkt und auf die tatsächlich auftretenden Fehlerbilder nicht zugeschnitten sind. Somit bietet sich die Erstellung von hauseigenen Fehlerbewertungslisten und Fehlerbildkatalogen an. Dieser Artikel erklärt, wie.
Die Erstellung von eignen Fehlerbildkatalogen und -listen läuft in folgenden Schritten ab:
- Schritt 1: Sammlung und Listung (potentieller) Produktfehler
- Schritt 2: Kategorisierung der (potentiellen) Produktfehler
- Schritt 3: Erstellung des Fehlerbildkatalogs
- Schritt 4: Erstellung der Fehlerbewertungsliste
Im Schritt 1 werden alle bereits aufgetretenen und alle potentiellen Fehlerbilder gesammelt. Als Beispiele für Quellen von Produktfehlern können Entwicklungsdokumente oder Risikoanalysen für neue Produkte und Herstellprozesse dienen, aber auch aufgetretene Fehlerbilder während der Entwicklung. Für etablierte Prozesse und Produkte kann man die Erfahrung aus manuellen Sortierungen, aus aufgetretenen Abweichungen sowie Rückmeldungen aus dem Markt (Complaints) nutzen.
Die Fehlerbilder werden systematisch aufgelistet und jedem Fehlerbild wird ein Severity Rank (Patientenrisiko durch das Fehlerbild) in einem Nummerierungssystem von beispielsweise 1 - 3 zugeordnet.
Im Schritt 2 wird anhand des individuellen Fehlerbildes basierend auf dem Severity Rank eine Entscheidung getroffen:
- Charge ist akzeptabel.
- Charge ist nicht akzeptabel.
- Acceptable Quality Level (AQL) nach DIN ISO 2859-1 zuordnen.
Zum Visual Management kann man die Fehlerbilder hierdurch in ein entsprechendes Ampelschema einordnen: Akzeptable Fehlerbilder kommen in einen grünen Bereich, AQL-belegte Fehlerbilder in einen gelben Bereich und nicht akzeptable Fehlerbilder in einen roten Bereich.
Im Schritt 3 wird basierend auf der Vorarbeit von Schritt 1 und 2 ein Fehlerbildkatalog erstellt. Dieser dient als Hilfestellung für die Mitarbeitenden an den Produktionslinien oder im Laborbereich zur Interpretation des Risikos von aufgetretenen Fehlerbildern. Die Fehlerbilder werden systematisch durchnummeriert, das Fehlerbild als Abbildung dargestellt, die Ampelfarbe dargestellt und eventuelle Folgemaßnahmen beschrieben. Diese Folgemaßnahmen können sein:
- Vernichtung der Charge
- 100 %- Kontrollen (vorzugsweise automatisiert)
- Auslösen einer Abweichung
Quellen von Abbildungen von Fehlerbildern können tatsächlich aufgetretene Fehler während der Produktion oder der Entwicklung sein. Alternativ können diese auch nachgestellt werden. Wichtig ist zusätzlich der Hinweis im Fehlerbildkatalog, neu auftretende, unbekannte Fehlerbilder über eine Abweichung zu bewerten und neu in den Katalog einzuordnen. Die Dokumente werden dann entsprechend versioniert.
Im Schritt 4 wird die Fehlerbewertungsliste für das Produkt erstellt. Hierbei handelt es sich um eine Checkliste, die von den Mitarbeitenden in der optischen Kontrolle ausgefüllt wird. Darin wird das Prüfergebnis eingetragen. In dieser Checkliste sind ebenfalls alle durchnummerierten Fehlerbilder mit der Prüfmethode und dem nötigen Stichprobenumfang, sowie den Akzeptanzkriterien aufgeführt. Die Folgemaßnahmen bei Nichterfüllung eines Akzeptanzkriteriums können der Fehlerbewertungsliste oder einer SOP zu den Dokumenten entnommen werden.
Abbildung 1 zeigt beispielhaft einen Ausschnitt einer Fehlerbewertungsliste in der Verpackung.
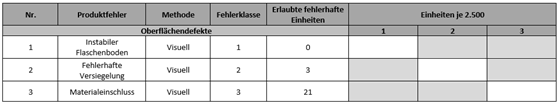
Abbildung 1
Zusammenfassung:
Fehlerbildkatalogen und Fehlerbewertungslisten kommen eine tragende Rolle in der Praxis des GMP-Alltags zu. Mit diesen Dokumenten wird die Qualität einer Charge dokumentiert und bewertet. Die Erfahrung zeigt: Diese Dokumente sollten durch den Betrieb selbst nach den beschriebenen Schritten 1 – 4 erstellt werden. So können der Katalog und die Fehlerbewertungsliste optimal auf den eigenen Prozess und das Produkt zugeschnitten werden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











