

Qualifizierung im Lebenszyklus einer Anlage
Auszug aus dem GMP:KnowHow Anlagenqualifizierung 2.0
6 Min. Lesezeit | von Dr. Julia Frej, René Mosinski
Erschienen im LOGFILE 04/2026
Von der ersten Risikoanalyse bis zur Stilllegung - der Lebenszyklus einer Anlage ist weit mehr als DQ, IQ, OQ und PQ. Wer Qualifizierung ganzheitlich denkt, schafft nicht nur Compliance, sondern legt den Grundstein für Qualität, Effizienz und nachhaltigen Projekterfolg.
Der Lebenszyklus einer Anlage beginnt mit der initialen Qualifizierung und erstreckt sich über die Implementierung, den Betrieb bis hin zur Außerbetriebnahme. Die abgeschlossene Qualifizierung weist dabei die Eignung von Gebäuden, Einrichtungen, Anlagen, Maschinen, Computern, Mitarbeitern und Lieferanten von Einsatzstoffen für den vorgesehenen Einsatzzweck nach. Die erfolgreiche Qualifizierung ist Voraussetzung für die Durchführung von Prozess- und Methodenvalidierung. Die Qualifizierung im Lebenszyklus einer Anlage kann wie folgt aussehen:
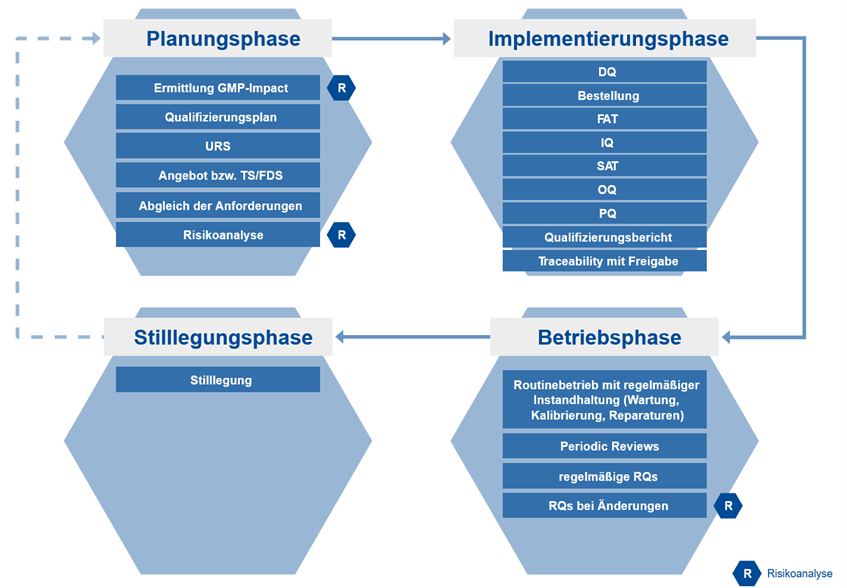
Bei einer neuen Anlage beginnt der Lebenszyklus bei der Planungsphase. Dort geht es darum, den GMP-Impact mit Hilfe einer Risikoanalyse zu ermitteln, den Qualifizierungsplan und das Lastenheft bzw. die URS zu erstellen, eventuell eine CSV-Systembeschreibung zu erstellen, das Angebot bzw. das Pflichtenheft (TS/FDS) einzuholen und mit den Anforderungen abzugleichen und schließlich wieder eine Risikoanalyse zu erstellen. Nach erfolgreichem Abschluss der Planungsphase, folgt die Implementierungsphase mit der Designqualifizierung (DQ), der Bestellung, dem Factory Acceptance Test (FAT), der Installationsqualifizierung (IQ), dem Site Acceptance Test (SAT), der Operational Qualification (OQ), der Performance Qualification (PQ), dem abschließenden Qualifizierungsbericht und der Tracebility-Matrix mit Freigabe. Anschließend folgt die Betriebsphase. Dort gibt es den Routinebetrieb mit regelmäßiger Instandhaltung (Wartung, Kalibrierung, Reparaturen), Periodic Reviews, regelmäßige Requalifizierungen bzw. Requalifizierungen bei Änderungen (inkl. Risikoanalyse) und sofern relevant: Überprüfung von Audittrail und Benutzerverwaltung im Rahmen der CSV. Wenn die Anlage außer Betrieb genommen werden soll, folgt schließlich die Stilllegung im Rahmen der Stilllegungsphase.
Planungsphase
Eine detaillierte Zeit-, Kosten- und Ressourcenplanung ist wesentlich für den erfolgreichen Verlauf des Gesamtprojektes. In der Praxis wird der Aufwand für Qualifizierungstätigkeiten regelmäßig unterschätzt. Infolgedessen ist der Zeitrahmen dafür nicht einzuhalten, und der Kostenrahmen wird gesprengt. Diese Planungen sollten bereits im Qualifizierungsmasterplan (QMP) enthalten sein.
Im Terminplan sollte vor allem für die frühe Projektphase genügend Zeit vorgesehen werden. Je später ein Fehler bemerkt wird, desto größer ist der Aufwand, ihn zu korrigieren. Wichtig ist eine Abschätzung, wie viele Testpunkte abgearbeitet werden müssen, wo dies erfolgt und ob ausreichend Kapazitäten in Labors und weiteren Fachabteilungen vorhanden sind. Auch gilt es Vorlaufzeiten (z. B. Rüstzeiten) einzuplanen.
Ein wichtiger Bestandteil aller Qualifizierungsaktivitäten sind GMP-Risikoanalysen. Sie dienen der Ermittlung potenzieller Risiken und deren Ursachen sowie der Festlegung geeigneter Maßnahmen. Mit GMP-Risikoanalysen können Umfang und Tiefe einer Qualifizierung festgelegt werden. Das Potential einer GMP-Risikoanalyse besteht dabei darin, die Anzahl der Tests oder Prüfungen zu reduzieren.
Für eine effektive Qualifizierung sollten alle Aktivitäten aus den Projektphasen für die Qualifizierung nutzbar sein, um Doppelarbeiten zu vermeiden. Dabei ist auf eine Gute Dokumentationspraxis zu achten.
Implementierungsphase
In der Designphase werden die Weichen für die Qualität einer Anlage gestellt, aber auch für den reibungslosen Ablauf des Gesamtprojektes. Die Designqualifizierung (DQ) umfasst die Dokumentation der Planungsphase einschließlich der Entscheidungsfindung. DQ-Dokumente beschreiben die Anforderungen des Auftraggebers zum Liefer- und Leistungsumfang (Lastenheft/URS) bzw. die Übereinkunft mit dem Auftragnehmer zur Realisierung und Abwicklung des Projektes (Pflichtenheft/TS/FDS).
Bei der Installationsqualifizierung (IQ) wird die Ausrüstung auf Übereinstimmung mit dem in der DQ (Designqualifizierung) erstellten Anforderungsprofil und auf ordnungsgemäße Installation geprüft. Die Installationsqualifizierung umfasst also nicht nur die korrekte Installation, sondern auch die Identifizierung des gesamten Lieferumfangs der Ausrüstung und die Bestätigung, dass sämtliche Bestandteile den im Pflichtenheft festgelegten Spezifikationen entsprechen. Ob die Inbetriebnahme vor oder nach der IQ erfolgen soll, hängt in erster Linie von der Art der Anlage ab. Um doppelte Arbeit und mehrmalige Durchführung identischer Prüfungen zu vermeiden, ist es unentbehrlich, den Zeitpunkt der Inbetriebnahme im Vorfeld zu planen. In vielen Fällen ist eine sichere Inbetriebnahme nur nach der IQ möglich. Hierzu zählen z. B. verfahrenstechnische Anlagen oder Wasseranlagen mit komplizierter Verrohrung.
Die Funktionsqualifizierung (OQ) dient dem Nachweis, dass die Anlage auf der Basis festgelegter Parameter und innerhalb definierter Grenzen funktioniert. Die OQ ist ein Prüfprozess, deshalb müssen die anzuwendenden Testverfahren sowie die Akzeptanzkriterien im Voraus bestimmt sein. Ebenso wie die Installationsqualifizierung gehört die OQ zu den standardmäßig durchzuführenden Projektphasen innerhalb der Qualifizierung. Je nach Art der Ausrüstung kann die Qualifizierung mit Abschluss der OQ beendet sein, da nicht alle Systeme zwingend einer Leistungsqualifizierung (PQ) unterworfen werden.
Im Rahmen der Leistungsqualifizierung (PQ) wird das richtige Zusammenspiel der bereits als Einzelbestandteile geprüften Ausrüstungsteile in größeren Einheiten wie z. B. Lüftungssystemen, Reinstwassersystemen, Abfüll- und Verpackungslinien geprüft.
Betriebsphase
Ein einmal erreichter Qualifizierungsstatus muss über den gesamten Lebenszyklus der Anlage bis hin zur Stilllegung garantiert werden. Dies erreicht man über regelmäßige Kalibrierungs-, Wartungs- und Instandhaltungsmaßnahmen, ein effektives Änderungsmanagement und regelmäßige Überprüfungen (Periodischer Review, PR). Änderungen können je nach ihrer Kritikalität und Auswirkung auf das Gesamtsystem zu einer Requalifizierung führen.
Die Gesamtheit dieser Maßnahmen führt dazu, dass die Komplexität der Anlage während ihres Lebenszyklus zunimmt. Dies erfordert eine gut organisierte und kontinuierlich gepflegte Dokumentation der Anlage und ihrer Historie. Auch das Ausfallrisiko nimmt mit der Betriebsdauer zu. Die damit verbundenen Risiken und Maßnahmen sind in einer gesonderten Risikoanalyse zu bewerten.
Stilllegungsphase
Der Lebenszyklus einer Anlage endet mit der Stilllegung. Dabei ist zu beachten, dass es sich bei der Stilllegung um eine Änderung handelt, die mit dem Änderungsmanagement unterliegt. Oftmals wird vor Stilllegung eines Systems nochmals eine Requalifizierung durchgeführt, um sicher zu stellen, dass die Anlage bis zum Schluss korrekt funktioniert hat.
Haben Sie Fragen oder Anregungen? Bitte kontaktieren Sie uns.redaktion@gmp-verlag.de











