

Abweichungsursachen als KPIs – Erhebung vs. Bewertung
7 Min. Lesezeit | von
Dr. Felix Kern und Liwa Schneider
Erschienen im LOGFILE Leitartikel 10/2022
Das Abweichungsmanagement spielt bei der Erhebung und beim Review von KPIs (Key Performance Indicators) bezüglich pharmazeutischer Produktions- und Qualitätsprozesse eine immer größere Rolle.
Speziell das Tracking der Abweichungsursache gibt Aufschlüsse darüber, wie stabil die jeweiligen Prozesse sind, wo sich Lücken befinden und wo mit CAPA-Maßnahmen gegengesteuert werden muss. Hierbei kann es jedoch zu Schwierigkeiten kommen, je nachdem wie diese Kennzahlen erhoben werden.
Generell gibt es eine große Vielfalt unterschiedlicher Ursachen für Abweichungen. Klassische Beispiele sind:
- Umweltbedingungen: Ein Sturm beschädigt die Lüftungsanlage für einen Produktionsbetrieb.
- Maschine: Ein Tablettierstempel bricht aufgrund eines Motorschadens.
- Human Error: Ein Mitarbeiter vergisst aufgrund einer Unachtsamkeit die Durchführung einer In-Prozess Kontrolle.
- Material: Bei der Wareneingangsprüfung finden sich Glaspartikel in einem Rohstoff.
- Methode: Eine Herstellungsanweisung ist zu unpräzise formuliert und ließ Raum für Interpretation oder wurde durch die Mitarbeiter nicht verstanden.
Die Herstellanweisung gibt Arbeitsschritte in unlogischer Reihenfolge vor. - Data Integrity: Es kommt während der Messung in einem Labor zu einem Systemabsturz mit Rohdatenverlust.
Besonders der KPI „Human Error“ – also der menschliche Fehler – nimmt an Bedeutung zu und wird in den Zielvorgaben vermehrt mit Obergrenzen belegt. Eine hohe Human Error Rate kann auf vieles hinweisen. Dies könnte beispielsweise sein:
- Eine oberflächliche Abweichungsbearbeitung. Es ist in vielen Fällen einfacher, die Ursache für eine Abweichung einem Mitarbeiterfehler zuzuordnen, als zeitaufwendig zu erforschen, was wirklich hinter dem Mitarbeiterfehler steckt. Das könnten beispielsweise Schulungsdefizite, Vorgabedefizite oder Systemdefizite sein.
- Ein systematisches Mitarbeiterproblem. Das kann beispielsweise Multi Tasking sein, was heißt, dass die Mitarbeiter zu viele Aufgaben parallel ausführen: Ein Mitarbeiter kümmert sich gleichzeitig um die Bedienung zweier Geräte: um die optische Kontrolle von Einheiten zu definierten Zeiträumen und um die Reinigung von Oberflächen.
- Es liegt ein tatsächlicher menschlicher Fehler vor. Dies könnten beispielsweise die bewusste Zuwiderhandlung gegen eine Anweisung oder auch eine Konzentrationsschwäche sein.
Allerdings muss auch darauf geachtet werden, wie die Human Error Rate erhoben wird. Hier stehen z. B. Absolutwerte oder Relativwerte zur Auswahl. Das bedeutet bei Absolutwerten, dass man von 100 Abweichungen 20 Abweichungen mit der Ursache „Human Error“ hat. Der Relativwert dazu wäre 20 %. Für die Nachverfolgung bietet sich der Absolutwert eher an als der Relativwert. Wieso ist das so? Hierzu im Nachfolgenden ein Beispiel:
Nehmen wir an, wir haben von 100 Abweichungen 50 mit der Ursache „Human Error“ und 50 mit der Ursache „Maschine“. Nun wird als Ziel (Relativwert) für die KPI Human Error 20 % festgelegt und es wird ein Projekt zur Reduktion der Human Error Rate aufgelegt. Gleichzeitig wird ein Projekt gestartet zur Stabilisierung der Herstellprozesse.
Durch das Human Error Reduktionsprojekt wird der Absolutwert an Abweichungen mit dieser Ursache von 50 auf 30 gesenkt. Ein riesiger Fortschritt. Gleichzeitig wird durch die Etablierung von modernen Maschinen und von modernen Prozessen zur Schonung dieser Maschinen die Anzahl an Abweichungen mit der Ursache „Maschine“ von 50 auf 10 reduziert.
Von 40 verbleibenden Abweichungen fallen 30 auf die Ursachenkategorie „Human Error“, was einem erhöhten Relativwert von 75 % entspricht. Das bedeutet, trotz einer respektablen Senkung der Anzahl der Abweichungen mit der Ursache „Human Error“ von 50 auf 30 Abweichungen (Absolutwert) ist der Relativwert von 50 % auf 75 % gestiegen (vgl. Tabelle 1).
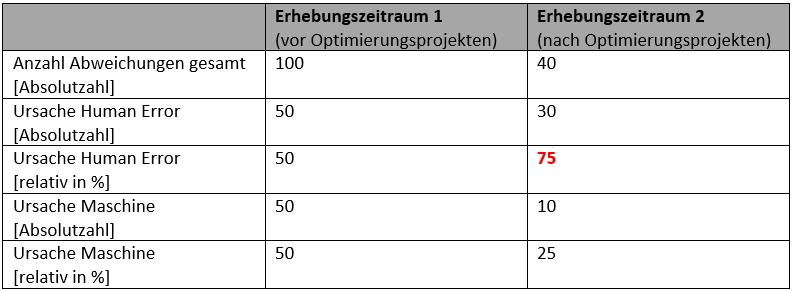
Tabelle 1
Dies betrachtet jedoch nur den Fall eines gleichbleibenden Produktionsvolumens. Bei einem steigenden Produktionsvolumen, beispielsweise von 100 auf 200 Chargen, ist davon auszugehen, dass auch der Absolutwert an Abweichungen steigen wird. Dies ist dann ebenfalls bei der Angabe der KPIs als Absolutwert zu berücksichtigen
Zusammenfassung
Das Tracking der Ursachenkategorien von Abweichungen nimmt in den KPIs eine immer größere Bedeutung ein. Besonders die Ursache „Human Error“ steht hier im Fokus. Dabei macht es mehr Sinn, diesen KPI nach dem Absolutwert zu tracken als nach dem Relativwert, wie wir im Beispiel anschaulich dargestellt haben. Um diese Zahlen richtig zu deuten und sinnvolle Maßnahmen daraus abzuleiten, muss man sich im Vorfeld überlegen, welche Faktoren zur korrekten Erhebung zu berücksichtigen sind. So können die KPIs letztendlich richtig bewertet werden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











