

GMP trifft Strahlenschutz – wie geht das zusammen?
2 Min. Lesezeit | von Andreas Nuhn
Erschienen im LOGFILE 05/2026
Die Herstellung von Radiopharmazeutika erfordert die gleichzeitige Einhaltung von EU-GMP-Leitfaden und Strahlenschutzverordnung, was zu Zielkonflikten zwischen Produktsicherheit und Personenschutz führen kann. Technische Lösungen wie Unterdrucksysteme und Bleizellen sowie eine frühzeitige Behördenabstimmung sind entscheidend für eine GMP- und strahlenschutzkonforme Umsetzung.
Bei der Herstellung von Radiopharmazeutika – unabhängig davon, ob sie für die Diagnostik oder für die Therapie eingesetzt werden – müssen Vorschriften aus zwei Rechtsbereichen eingehalten werden. Zum einen gelten die Produkte als Arzneimittel und müssen daher bei der Herstellung den GMP-Anforderungen hier insbesondere dem Anhang 1 des EU GMP-Leitfadens zur Herstellung von sterilen Arzneimitteln genügen. Der Anhang 3 des EU GMP-Leitfadens, der sich mit der Herstellung von Radiopharmazeutika befasst, verweist an vielen Stellen auf den Anhang 1 und gibt kaum spezifischen Anforderungen an technische Ausführungen zum Produktschutz. Zum anderen müssen die Anforderungen der Strahlenschutzverordnung eingehalten werden.
Diese Anforderungen aus den verschiedenen Dokumenten sind in manchen Bereichen widersprüchlich. Bei den GMP-Anforderungen steht die Patientensicherheit und damit die Produktsicherheit insbesondere, da es sich um nicht terminal sterilisierbare Parenteralia handelt, an erster Stelle. Aus Sicht des Strahlenschutzes müssen die technischen Maßnahmen jedoch sicherstellen, dass sowohl die Mitarbeiter im Strahlenschutzbereich als auch die Umgebung vor einer radioaktiven Kontamination essentiell geschützt werden (siehe Abbildung 1).
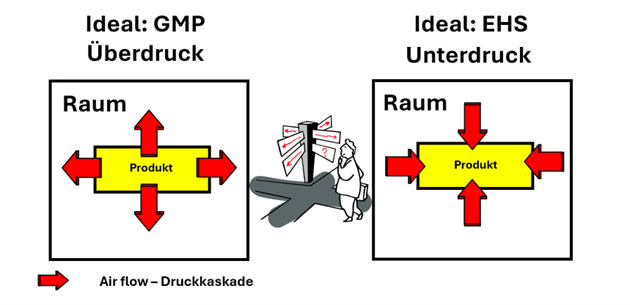
Abbildung 1 | Überdruck oder Unterdruck? (EHS: Umwelt-, Gesundheits-, Arbeitsschutz)
Bei der Projekt-Bearbeitung ist es daher wichtig, frühzeitig die Behördenvertreter in die technischen Diskussionen mit einzubinden, um teure Änderungen in der Detailplanung oder eventuell sogar bei bereits realisierten Installationen zu verhindern. Üblicherweise wird in Strahlenschutzbereichen mit absoluten Unterdrücken gearbeitet, um ein Austreten von Radioaktivität zu verhindern. Als Primärcontainment werden bei den Radiopharmazeutika Bleizellen sowohl für die Synthese als auch für die Abfüllung verwendet. Diese sind wie ein Isolator aufgebaut, jedoch kann der Operator nur über Manipulatoren (Greifer/Zangen) während der Herstellung in den Prozess eingreifen. Die Prozesse sind daher sehr stark automatisiert.
Um im Havariefall auch die Umgebung (umgebende Reinräume und weitere nicht klassifizierte Räume) vor radioaktiver Strahlung (belastete Luft) zu schützen, werden im nicht klassifizierten Bereich um die Reinräume (Herstellbereiche) zusätzlich Unterdruckzonen installiert. Hier ist dann insbesondere die Abstimmung mit der GMP-Überwachungsbehörde wichtig, da die klassische Kaskade – höchste Reinheitsklasse = höchster Druck – nicht gehalten werden kann (Bleizellen werden im absoluten Unterdruck gefahren). Der Reinraum (höchstes Druckniveau) wird dann im Allgemeinen über Druckspitzen oder Drucksenken vor Kontamination von außen geschützt. Diese Technik wird auch in der Herstellung mit kritischen Biologika (ab BSL 2) oder hochtoxischen Stoffen (ab OEB 4) verwendet.
Erfahren Sie mehr zu diesem spannenden Thema von Andreas Nuhn, D&B Pharmadesign GmbH, und Dr. Philipp Krapf vom Forschungszentrum Jülich am 24. März 2026 auf dem GMP-PharmaCongress in Wiesbaden. Ihr Vortrag hat den Titel “Case Study Forschungszentrum Jülich: The Challenge of Manufacturing Radiopharmaceuticals in Accordance with EU Annex 1”.
Mit Andreas Nuhn können Sie auch persönlich sprechen am 25./26. März 2026 auf den LOUNGES in Karlsruhe. D&B Pharmadesign finden Sie am Stand K4.3.
Haben Sie Fragen oder Anregungen? Bitte kontaktieren Sie uns.redaktion@gmp-verlag.de











