

Arzneimittelvermittlung – Fragen und Antworten
Hinweise und Interpretationen der dänischen Arzneimittelbehörde Laegemiddelstyrelsen
9 Min. Lesezeit | von Dr. Sabine Paris
Erschienen im LOGFILE Leitartikel 1/2023
Mit der Umsetzung der Fälschungsrichtlinie (2011/62/EU) in nationales Recht ist eine Anzeigepflicht für Arzneimittelvermittler (Broker) in die Arzneimittelgesetze der EU-Staaten aufgenommen worden.
Die dänische Arzneimittelbehörde Laegemiddelstyrelsen hat auf ihrer Website Fragen und Antworten zum Thema Arzneimittelvermittlung veröffentlicht. Der Leitartikel stellt einige der interessanten Fragen und Antworten zusammen.
Mit der Umsetzung der europäischen Fälschungsrichtlinie (2011/62/EU) in nationales Recht ist eine Anzeigepflicht für Arzneimittelvermittler (Broker) in die Arzneimittelgesetze der EU-Staaten aufgenommen worden.
Die dänische Arzneimittelbehörde Laegemiddelstyrelsen hat auf ihrer Website Fragen und Antworten zum Thema Arzneimittelvermittlung veröffentlicht.
Im Leitartikel habe ich einige interessante Fragen und Antworten der Dänen zusammengestellt und ins Deutsche übertragen. Die Antwort auf die erste Frage ist an die deutsche Definition im Arzneimittelgesetz angepasst worden.
Was ist Arzneimittelvermittlung?
Arzneimittelvermittlung ist jede berufs- oder gewerbsmäßig ausgeübte Tätigkeit von Personen, die, ohne Großhandel zu betreiben, selbstständig und im fremden Namen mit Arzneimitteln handeln, ohne tatsächliche Verfügungsgewalt über die Arzneimittel zu erlangen (§ 22a AMG).
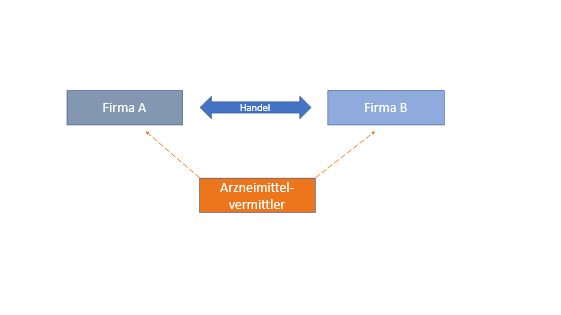
Abbildung 1 Arzneimittelvermittler
Gelten die Vorschriften für Vermittlungstätigkeiten auch für die Vermittlung von Arzneimitteln in und zwischen Drittländern (Ländern außerhalb der EU/des EWR)?
Die Vermittlung von Arzneimitteln gilt für Arzneimittel mit einer Zulassung in der EU/im EWR. Ein Unternehmen, das Arzneimittel mit einer Zulassung in der EU/im EWR an und/oder in Drittländer vermittelt, muss sich bei der zuständigen Arzneimittelbehörde registrieren lassen und unterliegt den europäischen bzw. den jeweils national festgelegten Regelungen.
Sollte ich mich als Arzneimittelvermittler registrieren lassen, wenn ich eine Genehmigung für den Großhandelsvertrieb habe?
Eine Genehmigung für den Großhandelsvertrieb deckt nur Großhandelsvertriebs-tätigkeiten ab (jede Form von Tätigkeiten im Zusammenhang mit dem Kauf, dem Verkauf, dem Empfang, der Lagerung und der Lieferung von Arzneimitteln innerhalb der EU/des EWR oder im Zusammenhang mit der Ausfuhr von Arzneimitteln in Drittländer).
Ein Unternehmen, das die Tätigkeiten des Großhandelsvertriebs und der Vermittlung ausübt, muss über eine Genehmigung für den Großhandel verfügen und sich als Vermittler von Arzneimitteln registrieren lassen. Dies gilt auch für Unternehmen, die über andere Genehmigungen verfügen, z.B. für die Herstellung und den Einzelhandelsvertrieb von Arzneimitteln.
Ist ein Verkäufer in Dänemark, der bei einem ausländischen Unternehmen beschäftigt ist, ein Vermittler oder ist eine dänische Tochtergesellschaft eines ausländischen Unternehmens ein Vermittler?
Im Allgemeinen nein. Ein Verkäufer, der bei einem ausländischen Unternehmen (Company A) angestellt ist und in Dänemark für Arzneimittel wirbt, gilt nicht als unabhängige Partei und fällt nicht unter den Begriff der Arzneimittelvermittlung. Dies gilt auch für Tochtergesellschaften, die im Namen eines ausländischen Unternehmens für Arzneimittel werben.
Solange der Verkäufer oder die Tochtergesellschaft keine Großhandelsvertriebstätigkeit ausüben (jede Form von Tätigkeit im Zusammenhang mit dem Kauf, dem Verkauf, der Entgegennahme, der Lagerung und der Lieferung von Arzneimitteln innerhalb der EU/des EWR oder im Zusammenhang mit der Ausfuhr von Arzneimitteln in Drittländer), müssen sie sich nicht als Vermittler registrieren lassen oder eine Genehmigung für den Großhandelsvertrieb in Dänemark besitzen.
Der Verkäufer oder die Tochtergesellschaft in Dänemark kann einen Auftrag entgegennehmen und ihn an Unternehmen A weiterleiten, was nicht als Vermittlung oder Großhandelsvertrieb angesehen wird.
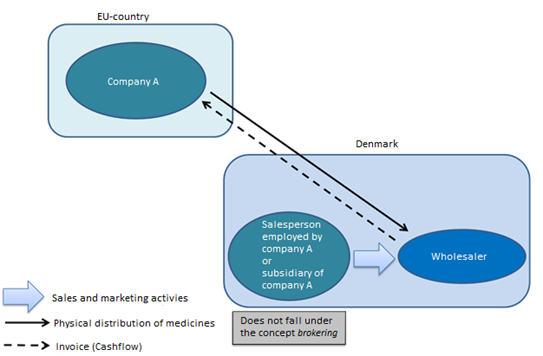
Abbildung 2 Szenario 1 (Quelle: Dänische Arzneimittelbehörde)
Wenn der Verkäufer oder die Tochtergesellschaft am Geldfluss beteiligt ist (Rechnungsstellung – Einkauf/Verkauf), ändert sich das Szenario, da dies als Tätigkeit für einen Großhändler betrachtet wird.
Dazu ist eine Genehmigung für den Großhandelsvertrieb in Dänemark erforderlich.
Fällt ein Unternehmen, das sich mit der Fakturierung, nicht aber mit der physischen Handhabung von Arzneimitteln befasst, unter den Begriff der Vermittlungstätigkeit, oder benötigt ein solches Unternehmen eine Genehmigung für den Großhandelsvertrieb?
Wenn ein Unternehmen am Geldfluss (Rechnungsstellung – Kauf/Verkauf) beteiligt ist, wird dies als eine Tätigkeit betrachtet, die unter den Begriff Großhandel fällt. Hierfür ist eine Genehmigung für den Großhandelsvertrieb in Dänemark erforderlich.
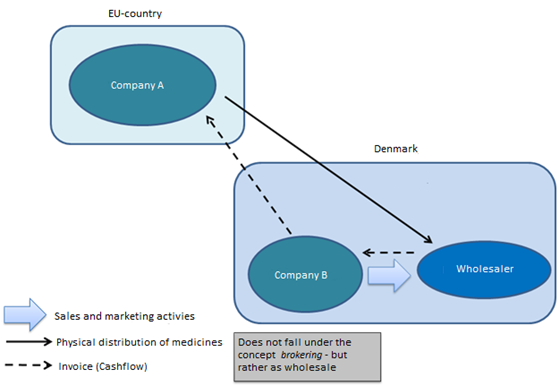
Abbildung 3 Szenario 2 (Quelle: Dänische Arzneimittelbehörde)
Hinweis für Deutschland: Eine regelmäßig aktualisierte Liste der den deutschen Behörden angezeigten Arzneimittelvermittler finden Sie auf der Website von PharmNet.Bund.
Quellen:
Deutsches Arzneimittelgesetz (GMP-BERATER Kapitel E.2)
Website der dänischen Arzenimittelbehörde Laedemiddelstyrelsen
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











