

Ausschaltung von Störfaktoren beim Bakterien-Endotoxin-Test (BET)
Auszug aus dem GMP-BERATER, Kapitel 12.H.6. - Kapitel 12.H.8
4 Min. Lesezeit | von Dr. Michael Rieth
Erschienen im LOGFILE Leitartikel 27/2021
Der empfindliche BET wird durch eine Reihe von Substanzen gestört bzw. beeinflusst. So können sowohl falsch-positive als auch falsch-negative Ergebnisse ermittelt werden.
Zu den Rohstoffen, die im Test interferieren, gehören unter anderem Salze mit zweiwertigen Kationen, Chelatbildner, Säuren, Antibiotika und Alkohole.
Beispielsweise rufen N-Methylglucamin, Lecithin und Detergenzien ein Enhancement (= Verstärkung) hervor.
Die Chelatbildner EDTA, EGTA, Heparin und Citrat und das Biozid m-Cresol wirken stark inhibitorisch.
Auch kann das Lysat durch Nicht-Endotoxin-Substanzen wie ß-1,3-D-Glucane, Trypsin, Chymotrypsin, Metronizadol und die Aminosäuren Threonin und Prolin aktiviert werden, sodass es zu falsch-positiven Reaktionen kommt.
Übliche Verfahren zur Ausschaltung der Störfaktoren sind
- Neutralisation
- Pufferung
- Verdünnung, maximal bis MVD
- Ultrafiltration, cut-off der UF-Membran: max. 20000 Dalton
- Dialyse
- Zentrifugation
- Hitzebehandlung
- Zugabe von Substanzen, die adsorbierte Endotoxine verdrängen
Wird die Ultrafiltration angewandt, empfiehlt Ph. Eur. asymmetrische Cellulosetriacetat-Membranen und rät von Polysulphon-Membranen ab. Der Filtereinsatz muss validiert werden, weil die Gefahr besteht, dass Cellulosederivate (ß-1,3-D-Glucane) abgegeben werden.
Durch Hitzebehandlung, z. B. 10 min bei 70°C, werden Serin-Protease-Inhibitoren in Plasma- und Seren-Proben inaktiviert. In der Laborpraxis haben Neutralisieren, Puffern oder Verdünnen die besten Erfolgsaussichten. Verwendet werden 0,1 M Kaliumphosphatpuffer pH 6,8 bis 7,0 und Tris-Puffer im Bereich 0,05 M bis 1 M, gegebenenfalls mit einem Zusatz von 0,5 M Magnesiumsulfat. Stark saure Proben können mit einer 0,2%igen Natriumacetatpuffer-Lösung pH 12 neutralisiert werden.
Die Verwendung des Lysats eines anderen Herstellers oder der Wechsel zu einer anderen Methode kann ebenfalls erfolgreich sein. So ist die chromogene Endpunktbestimmung weniger empfindlich gegenüber Interferenzen als die übrigen Methoden. Die turbidimetrische und die chromogene Methode liefern quantitative Ergebnisse bei hoher Empfindlichkeit, sodass geringe Endotoxinaktivitäten nachweisbar sind.
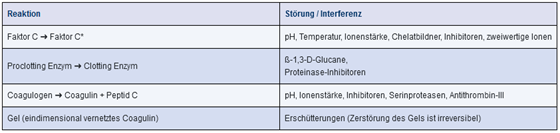
Abbildung 12.H-9 Störungen und Beeinflussungen der Reaktionen in der Gelbildungskaskade

Maskierung/Demaskierung
Das Phänomen der Maskierung von Endotoxinen führt zu falsch-negativen oder zu viel zu niedrigen Ergebnissen (Wiederfindung reproduzierbar kleiner als 50%). Die Endotoxine werden durch Interaktion mit bestimmten Substanzen verdeckt.
Petsch al. al. publizierten 1998 einen Artikel in der Fachzeitschrift Analytical Biochemistry zu diesem Phänomen1. Sie beschrieben, dass kationische Peptide und Proteine an die Endotoxine binden und somit die Reaktion mit dem Lysat erschweren. Im Test werden somit zu niedrige Endotoxin-Einheiten detektiert. Daher spricht man hier auch von “low endotoxin recovery – LER”. Dieser LER-Effekt tritt insbesondere dann auf, wenn in der Probe neben Tensiden wie Polysorbaten und Triton auch Phosphatsalze als Hilfsstoffe neben dem Wirkstoff enthalten sind. Chelatbildner wie Citrat, EDTA, EGTA sowie Substanzen wie Lecithin, die Liposomen bilden können, sind weitere Verursacher für den LER-Effekt. Da der Prozess der Maskierung reversibel ist, können verschiedene Verfahren zur Demaskierung versucht werden. So lässt sich der Maskierungskomplex mittels chaotroper Substanzen destabilisieren. Ein Demasking-Kit mit Namen Endo-RS® ist auf dem Markt.
Out of specification (OOS)
Die Vorgehensweise nach Auftreten eines OOS-Falls sollte in einer SOP beschrieben sein. OOS-Fälle müssen innerhalb einer definierten Zeit, z. B. 20 Arbeitstage, abgeschlossen sein. Das OOS-Verfahren wird angestoßen, wenn die Ursache für das außerhalb der Spezifikation liegende Ergebnis nicht unmittelbar als Analysen- oder Gerätefehler bzw. technische Störung erkannt wird. Der Laborleiter erstellt vor Beginn weiterer Untersuchungen einen Prüfplan und legt, wenn möglich, die Akzeptanzkriterien fest. Ein Nachmusterzug soll nur dann geschehen, wenn der Verdacht besteht, dass das Originalmuster nicht repräsentativ oder inhomogen ist, oder wenn es aufgebraucht wurde. Unter Umständen kann es sinnvoll sein, dass die Nachanalyse durch einen anderen Laboranten ausgeführt wird.
Die Wiederholungsprüfung soll so angelegt sein, dass das Ergebnis mehr Gewicht als das der Originalanalyse hat. Dazu kann z. B. ein Muster einer bereits erfolgreich geprüften Charge parallel untersucht werden. In der SOP muss auch festgelegt sein, ob ein statistischer Ausreißertest zugelassen wird.
Mögliche Ursachen für Ausreißer sind:
- Ablesefehler
- Fehler bei der Messwertübertragung
- Verwechslungen
- Beschädigung des Messobjekts
Als Ausreißertests kommen der Grubbs-Test (wenn 1 Messwert verdächtig ist, siehe DIN 32645 und 38402) oder graphische Verfahren wie Box-Plot (siehe dazu Statistik-Software wie Minitab 15®) zur Anwendung.
Ursachen für OOS-Fälle:
- Probe beim Musterzug/-transport kontaminiert
- Laborkontamination (Sekundärkontamination)
- Probe ist tatsächlich Endotoxin-belastet („Endoburden“)
- falsch positiv durch ß-1,3-D-Glucane, Trypsin, Chymotrypsin o. a.
Zu 1) Probennehmer oder Probengefäß nicht Endotoxin-frei (hitzebeständige Geräte und Gefäße aus Glas oder Metall werden gemäß Ph. Eur. – Kapitel 2.6.14 im Heissluftsterilisator nach einem validierten Verfahren bei mindestens 250°C und einer Dauer von mindestens 30 min entpyrogenisiert). Eine weitere Ursache für eine Endotoxin-Kontamination kann die Probennahme in feuchter Umgebung, z. B. nach einer Reinigung, sein. Beim Probentransport können Kontaminationen passieren, wenn das Probengefäß beschädigt wird oder wenn es nicht richtig verschlossen ist.
Zu 2) Indizien für eine Laborkontamination sind die positiv ausgefallene Negativkontrolle und ungewöhnlich hohe Endotoxinwerte in allen oder nahezu allen Proben.
Zu 3) Natürlich kann die Probe auch tatsächlich mit Endotoxinen kontaminiert sein. Daran ist vor allem zu denken, wenn die zu prüfende Substanz aus natürlichem Material (tierischer, pflanzlicher oder mineralischer Ursprung) gewonnen wurde.
Zu 4) Serinproteasen wie Trypsin und Chymotrypsin können in die Aktivierungskaskade eingreifen und so ein falsch-positives Ergebnis vortäuschen. ß-1,3-D-Glucane aktivieren den Faktor G-Weg, was zu einer Gelierung oder Trübung führt.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











