

Auswahlkriterien für ein geeignetes eDMS
Ein Auszug aus dem GMP-BERATER, Kapitel 15.G.6, Auswahlkriterien für ein geeignetes eDMS
7 Min. Lesezeit | von Thilo Gukelberger
Erschienen im LOGFILE Leitartikel 14/2021
Während früher IT-Systeme meist direkt in den Firmen auf eigener IT-Infrastruktur installiert wurden, geht heute der Trend zu cloudbasierten Systemen. Der Betrieb eines validierungspflichtigen Systems in einer Cloud bedarf der zusätzlichen Klärung und Abstimmung mit dem Cloudbetreiber.
Auch stellt sich die Frage, ob man ein einheitliches eDMS für GxP- und non-GxP-Bereiche einführt oder für diese Bereiche getrennte Systeme einsetzt. Details dazu lesen Sie heute in einem Auszug aus dem weltweit größten GMP-Wissensportal, dem GMP-BERATER.
Betriebsform
Im Folgenden werden unterschiedliche Betriebsformen für ein elektronisches Dokumentenmanagementsystem (eDMS) beschrieben. Aus GMP-Sicht ist dabei zu berücksichtigen, dass die Räume, in denen das eDMS betrieben wird, in der Betriebserlaubnis aufgeführt sein müssen. Die Nutzung eines externen Rechenzentrums, z. B. beim ASP-Betrieb oder Betrieb des eDMS in der Cloud, muss im Site Master File, insbesondere in den Abschnitten 4 und 5, dokumentiert werden. Solche extern gehosteten Lösungen stellen somit aus GxP-Sicht besondere Anforderungen an den Betrieb und den externen Dienstleister.
On-Premises
Ein eDMS als On-Premises-Lösung ist die klassische Betriebsform. Hierbei wird das IT-System beim pharmazeutischen Unternehmen (Kunde) installiert. Das Programm und die verwalteten Daten liegen somit auf den Servern des Kunden. Somit hat der Kunde die volle Kontrolle über das System. Jedoch muss er auch die Hardware (Server), den Speicher, üblicherweise das Betriebssystem (meist Microsoft Windows Server) und die Datenbank (z. B. Microsoft SQL-Server oder Oracle) bereitstellen. Auch obliegen dem Kunden Arbeiten wie die Administration des Systems und die Datensicherung.
Application Service Providing (ASP)
Bereits seit vielen Jahren ist das Application Service Providing ein häufig genutztes Betriebsmodell für IT-Lösungen. Hierbei übernimmt der Anbieter den kompletten Betrieb der IT-Lösung. Die Software kann auf Hardware des Kunden vor Ort, auf Hardware des Anbieters oder eines externen Rechenzentrums betrieben werden. Entscheidend ist, dass der Kunde nur Anwender des IT-Systems ist und sich nicht um die IT-Administration kümmern muss. Jedem Kunden wird hier eine eigene Anwendungsumgebung bereitgestellt. Auf spezielle Kundenanforderungen kann daher sehr flexibel reagiert werden. Für die Software selbst wird dabei in der Regel kein Kaufpreis entrichtet sondern der Kunde mietet das Nutzungsrecht für die Software.
Cloud
Seit Mitte der 2010er Jahre gewinnt Cloudcomputing mehr und mehr an Bedeutung. Hierbei sind jedoch verschiedene Varianten zu unterscheiden.
Bei der public cloud wird das IT-System auf Hardware des Cloud-Anbieters betrieben. Mehrere Kunden teilen sich in der Regel ein gemeinsames IT-System. Die Einspielung von Softwareupdates führt der Anbieter i. d. R. ohne explizite Einwilligung der Kunden durch. Oftmals ist nicht transparent, wo die Server stehen, auf denen die Daten gespeichert bzw. repliziert werden. Für validierungspflichtige Systeme ergeben sich hier gravierende Herausforderungen für den Kunden, der aus GxP-Sicht ja verantwortlich für den Betrieb des Gesamtsystems bleibt.
Die private cloud wird nur für einen einzelnen Kunden betrieben. Der Kunde hat somit die Kontrolle über sein System. Die private cloud ähnelt damit dem Application Service Providing, wobei bei der private cloud die IT-Infrastruktur im Vordergrund steht, beim ASP hingegen die IT-Anwendung.
Für das GxP-Umfeld könnte die sogenannte community cloud interessant sein. Bei diesem Modell teilen sich mehrere Kunden mit sehr ähnlichen Anforderungen eine gemeinsame, über Internetdienste bereitgestellte Anwendung. Kunden aus dem GxP-Umfeld haben dieselben regulatorischen Anforderungen. Wenn der Betreiber der community cloud über entsprechende Expertise im GxP-Umfeld verfügt, dann können die Anforderungen der Validierung und Revalidierung des IT-Systems konform zu den Regularien umgesetzt werden und die Kosten für diese aufwändigen Arbeiten auf mehrere Kunden aufgeteilt werden. Auch bei einem solchen gemeinsam genutzten System ist es aus GMP-Sicht erforderlich, dass jeder Kunde seine individuellen Anforderungen in einer URS/DS festhält und die Einhaltung dieser Spezifikation auch nachweist. So können sich die Anforderungen z. B. in Bezug auf Aufbewahrungsfristen, Anonymisierung oder Zugriffsrechte durchaus von Nutzer zu Nutzer unterscheiden, auch wenn alle die gleiche Cloud-Lösung nutzen.
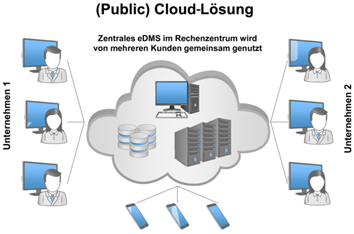

Abbildung 15.G-5 Schematische Darstellung einer Public Cloud-Lösung
Anwendungsbereich
Getrennte Systeme für GxP- und non-GxP-Anwendungen
Die Qualifizierung und Validierung eines IT-Systems ist mit erheblichem Aufwand verbunden. Die erstmalige Validierung ist aber nur ein Aspekt. Von Zeit zu Zeit erfordern IT-Systeme die Einspielung von Updates. Diese unterliegen dem Change Control und erfordern ggf. eine Revalidierung des Systems. Bei Betrieb eines validierten IT-Systems sollte man daher bestrebt sein, so viele Änderungen wie nötig, aber nur so wenige Änderungen wie möglich einzuspielen. Soll das eDMS nicht nur GxP-relevante Dokumente verwalten, dann kann die Notwendigkeit der Einspielung von Änderungen für den non-GxP-Bereich unerwünschte Zusatzarbeit nach sich ziehen. Daher tendieren einige Unternehmen zur Trennung und dem Betrieb separierter eDMS. Stammen die eingesetzten IT-Lösungen nicht vom selben Anbieter, dann bedeutet der Betrieb zweier unterschiedlicher Systeme jedoch höhere Kosten, sowohl intern als auch extern. Auch bedeutet es für den/die Anwender:in, der mit Dokumenten aus beiden Welten arbeitet, sich in zwei unterschiedlichen Systemen bewegen zu müssen.
Einheitliches System für GxP- und non-GxP-Anwendungen
Wünschenswert ist aus Kundensicht der Betrieb eines einzigen und einheitlichen eDMS für sämtliche Dokumente – GxP und non-GxP. Solche Systeme sind jedoch selten am Markt zu finden. Da die Anforderungen der unterschiedlichen Bereiche sehr unterschiedlich sind, konzentrieren sich die meisten Anbieter auf eine der beiden Welten. Hat man sich jedoch für ein eDMS entschieden, mit dem sämtliche Dokumente des Unternehmens verwaltet werden, dann muss der Anbieter vor geplanten Updates eine Risikobewertung für GxP-relevante Anwendungsfälle liefern, denn nur diese sind für die Revalidierung relevant.
Der große Vorteil eines einzigen eDMS für sämtliche Unternehmensdokumente liegt in den geringeren Kosten und dem komfortableren Arbeiten für Mitarbeitende, die mit Dokumenten aus beiden Welten arbeiten müssen.
Beide Optionen – sowohl getrennte als auch gemeinsame Systeme – haben Vor- und Nachteile. In Abbildung 15.G-6 sind einige Entscheidungskriterien aufgeführt.


Abbildung 15.G-6 Argumente für die Auswahl gemeinsamer oder getrennter Systeme
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











