

CEP-Verfahren: Vor- und Nachteile
Ein Auszug aus dem GMP-BERATER, Kapitel 20.C.2, Certificate of Suitability of Monographs of the European Pharmacopoeia (CEP)
7 Min. Lesezeit | von Prof. Dr. Markus Veit
Erschienen im LOGFILE Leitartikel 18/2021
Ablauf und Zuständigkeiten
Für die Beantragung eines CEPs reicht der Wirkstoffhersteller die Dokumentation des Kapitels 3.2.S beim European Directory for the Quality of Medicines and Healthcare (EDQM) ein.
Es handelt sich dabei um eine Behörde des Europarates, die das Europäische Arzneibuch herausgibt. Mit der Einreichung beantragt der Wirkstoffhersteller die Ausstellung eines CEPs. Der Antragsteller verpflichtet sich dabei auch zur Teilnahme am Inspektionsprogramm des EDQM, mit dem alle Antragsteller formal der GMP-Überwachung durch das EDQM unterliegen.
Die Prüfung der eingereichten Wirkstoffdokumentation erfolgt beim EDQM durch einen Pool an Assessoren, die aus unterschiedlichen Bereichen kommen können. In der Regel erfolgt die Beurteilung durch einen einzelnen Assessor, das Ergebnis wird dann durch einen zweiten kontrolliert. Die eingereichte Dokumentation verbleibt beim EDQM und wird von den nationalen Zulassungsbehörden in der Regel nicht eingesehen, wenngleich dies prinzipiell möglich wäre.
Nach der Erteilung eines CEPs wird dieses in einer Datenbank des EDQM gelistet. Damit ist die Information, für welche Wirkstoffe welcher Hersteller CEPs erteilt wurden, allgemein zugänglich.
Im Zulassungsverfahren für ein Arzneimittel fragt der Antragsteller dann den Halter des CEP (also seinen Wirkstofflieferanten) an, ob er auf das CEP Bezug nehmen darf. Daraufhin stellt der Wirkstoffhersteller eine sogenannte „Written Confirmation“ aus, in der er versichert, den Antragssteller nach Erteilen der Zulassung über alle Änderungen zu informieren. Diese „Written Confirmation“ und das eigentliche CEP werden mit dem Zulassungsdossier eingereicht (siehe Abbildung 1).
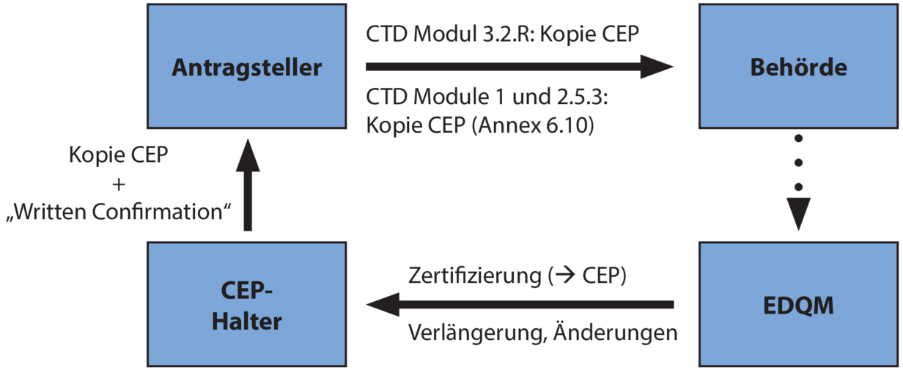
Abbildung 1 CEP-Verfahren
Zweck des CEP-Verfahrens
Die Regelung zu dem Verfahren findet sich im Anhang I der Europaratsresolution „AP-CSP (07) 1 Zertifizierung der Eignung zu den Monographien des Europäischen Arzneibuchs“. Das auf dieser Resolution basierende Verfahren wurde 1999 eingerichtet, um sicherzustellen, dass die Qualität des Wirkstoffs mit den in der Arzneibuchmonographie enthaltenen Kontrollmethoden angemessen überprüft werden kann. Das gilt insbesondere für die Prüfung auf Reinheit, also den Gehalt an Verunreinigungen. Diese können aus der Herstellung des Wirkstoffs stammen oder durch Abbau entstehen. Sollten die in der Stoffmonographie enthalten Kontrollverfahren im Lichte der spezifischen Synthese des Wirkstoffs nicht ausreichen, können Beschreibungen für zusätzliche Kontrollverfahren eingereicht werden. Diese werden dann in das CEP übernommen und müssen im Rahmen der Chargenfreigabe für diesen spezifischen Wirkstoff verpflichtend geprüft werden. Es obliegt der Verantwortung des Wirkstoffherstellers, dazu eine umfassende Bewertung möglicher Verunreinigungen vorzunehmen und diese ggf. angemessen zu kontrollieren.
Anerkennung von CEPs bei der Zulassung von Arzneimitteln
CEPs werden von den 38 Mitgliedstaaten der Arzneibuch-Konvention sowie einer Reihe anderer Länder (z. B. Australien, Südafrika, Saudi-Arabien) anerkannt. Diese Anerkennung bedeutet, dass die Unterlagen zur Qualität der Wirkstoffe im Rahmen von Arzneimittel-Zulassungsverfahren von den zulassenden nationalen Behörden nicht mehr geprüft werden müssen, sondern die Prüfung durch das EDQM anerkannt wird.
Vorteile des CEP-Verfahrens
Das CEP-Verfahren hat eine Reihe von Vorteilen für alle beteiligten Parteien:
- Es schafft die Sicherheit, dass generische Wirkstoffe auf der Basis der Arzneibuchmonographie geprüft werden können – auch wenn die Herstellung von der Synthese des Originalherstellers abweicht. Anders wäre das Arzneibuch weitgehend wertlos. Die Möglichkeit, die Prüfung gemäß der Arzneibuchmonographie durchzuführen, entlastet den Arzneimittelhersteller und sorgt für eine bessere Vergleichbarkeit.
- Die Zulassungsbehörden in Europa sind mit dem Verfahren auch nicht unbedingt unglücklich, spart es doch bei ihnen die Ressourcen, die für die Beurteilung der Unterlagen zum Wirkstoff erforderlich wären.
- Das Verfahren wahrt auch die Interessen der Wirkstoffhersteller, denn es garantiert einen umfassenden Schutz der eingereichten Unterlagen und damit des geistigen Eigentums des Wirkstoffherstellers.
- Auch die Pharmazeutischen Unternehmer, bzw. Zulassungsinhaber, respektive Inverkehrbringer sind nicht unglücklich mit dem Verfahren, denn sie können damit weite Teile der Verantwortung für eine inhaltlich mit den Anforderungen übereinstimmende Dokumentation der Qualität der Wirkstoffe und ihrer Herstellung an das EDQM abgeben.
Nachteile und Risiken des CEP-Verfahrens
Alle „Nutzer“ von CEPs müssen sich sicher sein, dass sowohl beim Wirkstoffhersteller als auch beim EDQM die Hausaufgaben lege artis gemacht werden. Sollten sie daran Zweifel haben oder einfach aus Gründen der Sorgfaltspflicht einen Einblick in die Unterlagen verlangen, haben sie darauf keinen Rechtsanspruch. Einen solchen Anspruch haben nur die Zulassungsbehörden. Nach den Erfahrungen des Autors nutzen die Behörden diesen Anspruch jedoch nur selten und überlassen die Beurteilung der Qualität des Wirkstoffs den Assessoren des EDQMs – nicht zuletzt sicher auch wegen mangelnder Ressourcen.
Im Rahmen der Zertifizierung hat das EDQM die Möglichkeit, wirkstoffspezifische Inspektionen bei den Wirkstoffherstellern (auch in außereuropäischen Ländern) durchzuführen und zu überprüfen, ob die eingereichten Unterlagen der gelebten Wirklichkeit entsprechen. Solche Inspektionen finden jedoch nur risikobasiert statt, wenn es Anhaltspunkte zu einer Non-Compliance gibt.
Der „Valsartan-Fall“ eignet sich gut, das bestehende System kritisch zu hinterfragen: Der chinesische Hersteller Zhejiang Huahai Pharmaceutical Co. Ltd hatte für das von ihm vertriebene Valsartan ein CEP, das 2016 vom EDQM verlängert wurde. Offenbar war dabei den Assessoren des EDQM bei der Beurteilung der eingereichten Unterlagen das Risiko des Entstehens von Nitrosaminen im Herstellungsprozess nicht aufgefallen, obwohl die international gültige ICH-Leitlinie M7 eine umfassende Bewertung des Synthesewegs hinsichtlich möglicher mutagener bzw. genotoxischer Verunreinigungen für neue oder geänderte Prozesse auch für generische Wirkstoffe zwingend vorschreibt.
Fazit
Im „Valsartan-Fall“ hatten weder die zulassenden oder überwachenden Behörden noch die Inverkehrbringer die Informationen, die eine Erkennung des Risikos ermöglicht hätten. Der Inverkehrbringer hätte noch nicht einmal die Möglichkeit gehabt, diese Informationen einzufordern. Er hat lediglich die Pflicht, den Wirkstoffhersteller zu auditieren und sich systemisch (nicht wirkstoffspezifisch) Gewissheit zu verschaffen, dass der Wirkstoff entsprechend der europäischen GMP-Vorgaben hergestellt wird.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











