

Die nachhaltigere Methode - Erzeugung von WFI mit Membrantechnik
Ein Auszug aus dem GMP-BERATER, Kapitel 5.B.11, Erzeugung von WFI mit Membrantechnik
8 Min. Lesezeit | von Dr. Herbert Bendlin und Fritz Röder
Erschienen im LOGFILE 16/2022
Diese wurden bisher zur Erzeugung von Hochgereinigtem Wasser (HPW) eingesetzt. Diese Wasserqualität, die sich bis dato nur durch die Herstellungsmethode von WFI unterschied, ist seit April 2019 nicht mehr im Europäischen Arzneibuch enthalten.
Die Erzeugung von WFI mittels Membrantechnik wird auch als „kalte“ WFI-Herstellung bezeichnet. Mittlerweile wurden zahlreiche Anlagen seitens der Industrie bei den Anlagenbauern bestellt, in Betrieb genommen und qualifiziert. Die Anlagen erzeugen sicher und zuverlässig WFI.
Zu bedenken ist in diesem Zusammenhang, dass die Anwendungsrisiken für Arzneimittel, für die man WFI verwendet, höher sind als für Arzneimittel, die bisher mit HPW hergestellt wurden, beispielsweise Ophthalmika (Produkte zur Anwendung am Auge). Arzneimittel, die parenteral verabreicht werden und WFI enthalten, verteilen sich direkt im Blutkreislauf. Es gibt keinerlei Barriere mehr, die eventuelle Verunreinigungen zurückhalten könnte. Allein durch die Anwendung des Arzneimittels (Applikationsweg) ergeben sich also höhere Risiken und somit Anforderungen an die Technik. Eine Qualifizierung von WFI-Membrananlagen muss daher mit entsprechender Sorgfalt durchgeführt werden. Die Risiken der kalten Herstellung werden seitens der Hersteller, Betreiber und Behörden noch unterschiedlich beurteilt. Auch spielt die Betriebsweise und Überwachung beim Anwender eine größere Rolle als sie bei der rein technischen Bewertung zum Tragen kommt.
Wenn man auf Membrantechnik zur WFI-Erzeugung umstellen möchte, ist die zuständige Überwachungsbehörde darüber zu informieren. Dies ist eine Anforderung der Ph. Eur.-Monographie 0169.
Der wesentliche Vorteil der Membrantechnik liegt in den geringeren Kosten: bisher hatte man ein System für Gereinigtes Wasser und schaltete eine zweite Anlage zur Verdampfung nach, um WFI zu erhalten. Man brauchte auch zwei Lager- und Verteilsysteme. Das alles kann nun entfallen. Man stellt nur noch WFI per Membranverfahren her und benutzt dieses für alle Anwendungen. So entfallen die Anschaffungs-, Betriebs- und Energiekosten für das heiße System sowie die zweite Verteilung im Gebäude.
Ein weiterer wesentlicher Grund für die Anschaffung eines kalten WFI-Systems dürfte in Zukunft der Faktor „CO2-Fußabdruck“ bzw. „Nachhaltigkeit“ sein. Große Firmen haben sich durchweg Ziele gesetzt, in einem Zeitraum von fünf bis zwanzig Jahren klimaneutral zu werden. Die Verwendung von Dampf in jeglicher Form trägt wesentlich zur CO2-Bilanz eines jeden Unternehmens bei. Um also jene Klimaziele zu erreichen, ist damit zu rechnen, dass auf Dauer kalte WFI-Systeme zunehmend häufiger installiert werden.
Der Aufbau einer Anlage zur Herstellung von HPW-Wasser oder kaltem WFI ist in folgender Abbildung dargestellt.
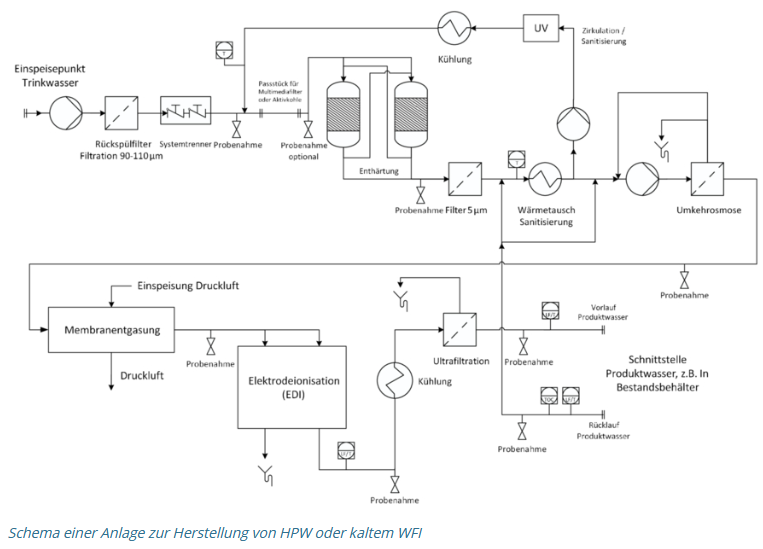
Aufgrund der erhöhten Risiken bei der parenteralen Anwendung weist die aktuell gültige Leitlinie der EMA auf verschiedene zusätzliche Kontrolleinrichtungen hin, die bis dato bei HPW nicht notwendige Praxis waren. Darunter fallen zum Beispiel:
- Der Einsatz von Schnellmethoden für Keimzahl- und Endotoxinbestimmung (in Verbindung mit den konventionellen Bestimmungsmethoden gemäß Monographie)
- Der Einsatz kombinierter Sanitisierungsverfahren mittels Hitze UND Chemikalien
- Erhöhter Wartungs- und Überwachungsaufwand (Austausch von RO-Membranen/Beprobungsfrequenzen)

Weitere geforderte Maßnahmen, die aber auch für HPW bereits galten, sind z. B.:
- Nachweis der Integrität der UF-Module
- Analyse der Rohdaten mittels statistischer Tools zur Bewertung der Warn- und Aktionsgrenzen
- eine ganzheitliche Prozesskontrollstrategie
Erste Anlagen zur WFI-Herstellung mit Umkehrosmose sind bereits in Betrieb gegangen und sind qualifiziert. Die Reaktionen der Behörden in Europa dazu fielen bisher sehr unterschiedlich aus (von „keine Reaktion“ bis zu „Vor-Ort-Besuch“). Es lässt sich feststellen, dass hier kein europaweit standardisierter Rechtsvorgang bei Behörden existiert. Dennoch dürfte die behördliche Akzeptanz auf Dauer weiter steigen, da diese Systeme jetzt schon zuverlässig funktionieren und es mutmaßlich aufgrund des Themas „Nachhaltigkeit“ mehr und mehr Systeme in Zukunft geben wird.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











