

Die Risiken von Datenintegritätsfehlern erkennen
7 Min. Lesezeit | von Dr. Siegfried Schmitt
Erschienen im LOGFILE 03/2022
Seit den frühesten Aufzeichnungen gibt es Integritätsprobleme. Eines der ersten wurde bereits 300 Jahre v. Chr. aufgezeichnet.
Da gab es den griechischen Kaufmann Hegestratos. Hegestratos nahm einen Bodmereibrief über eine große Summe auf (das ist ein Schiffspfandbrief, bei dem Schiff und Ladung als Sicherheit für das Darlehen dienen). Dabei verfällt die Sicherheit, falls das Darlehen nicht zurückgezahlt wird. Hegestratos heckte einen Plan aus, nachdem er das leere Schiff versenken, die Ladung (bestehend aus Mais) und das Darlehen aber behalten wollte. Unglücklicherweise wurde Hegestratos aber von seiner Mannschaft auf frischer Tat ertappt. Er ertrank auf der Flucht. Sein Mitverschwörer wurde gefasst und angeklagt (Beattie, 2015).
Das Management sollte sich darüber im Klaren sein, dass es in regulierten Branchen Integritätsverletzungen gibt, seit es Gesetze und Vorschriften gibt. Die Vergangenheit bietet zahllose bemerkenswerte Beispiele an Integritätsverletzungen, darunter einige mannigfach publizierte Vorfälle. Es gibt keinen Grund anzunehmen, dass Integritätsverletzungen jemals von alleine verschwinden werden. Das verantwortliche Management sollte also davon ausgehen, dass Integritätsverletzungen jederzeit in sämtlichen Bereichen entstehen können. Deshalb sollte das verantwortliche Management seine Pflicht tun und wirksame Kontrollen (für Systeme und Prozesse) etablieren, um Fehler zu verhüten bzw. zu entdecken. Falls solche Fehler passieren, sollten die Ursachen behoben werden, um eine Wiederholung zu vermeiden.
Chester Bowles (1901–1986), ein früherer Gouverneur von Connecticut, beschreibt seine Compliancetheorie wie folgt:
„20 % einer regulierten Bevölkerung erfüllen ohne nachzudenken sämtliche Vorschriften. 5 % versuchen sie zu vermeiden und die verbleibenden 75 % werden diese erfüllen, solange sie glauben, dass die 5 % erwischt und bestraft werden.” (Kelliher, 2007)
Ein vernünftiger Managementansatz für die Überprüfung von GMP-Aufzeichnungen lautet: „Vertrauen ist gut, Kontrolle ist besser”. Betrachten wir also die übliche Herangehensweise des Managements zur Überprüfung der Protokolle der Chargenfertigung. Viele glauben, was in Formularen, Aufzeichnungen und Berichten geschrieben steht, ohne routinemäßig die Genauigkeit und Vollständigkeit zu prüfen, indem sie Daten und Informationen mit den Originalaufzeichnungen vergleichen. Einige unterlassen eigene Schritte, um die Genauigkeit oder Vollständigkeit des Geschriebenen zu bestätigen, da sie den Mitarbeitern „vertrauen“ und sie für ehrlich halten. Andere verlassen sich darauf, dass ihr Qualitätssystem wirksam ist, und es keinen Grund gibt, anzunehmen, dass die Daten und Informationen nicht wiedergeben, was wirklich passiert ist.
Integritätsverletzungen passieren jedoch immer dann, wenn man sie am wenigstens erwartet. Selbst falls gut konzipierte Programmen und Kontrollen für das Datenintegritätsmanagement vorhanden sind, kann jede Einzelperson (oder Gruppe) absichtlich von geltenden Anforderungen abweichen. Auch können wohlmeinende Einzelpersonen unabsichtliche Fehler beim Sammeln, Überprüfen, Berichten und Aufbewahren von GMP-Daten machen. Somit ist es erforderlich, dass das Management seinerseits fortlaufend die Datenintegritäts-Kontrollprozesse überwacht, um sicherzustellen, dass sämtliche Daten korrekt, wahr und vollständig sind und etwaige Fehler entdeckt werden. Das Management muss ständig wachsam bleiben und nach unabsichtlichen Fehlern, wie z.B. Irrtümern oder Auslassungen, Ausschau halten. Es muss außerdem auf Anzeichen für absichtliches Fehlverhalten, wie Betrug und Fälschungen, achten.
Das Management muss stets wachsam bleiben und die Gründe für beabsichtigte Datenintegritätsfehler (Betrug oder Fälschungen) kennen und verstehen. Joseph T. Wells sagte (Wells, 2001): „Es ist entweder Gier oder Not.” David Chesney drückte es so aus (Chesney, 2015): „Es geht entweder darum, Ärger zu vermeiden oder Gewinn zu machen.” Abbildung 1 zeigt, warum Einzelpersonen willentlich Datenintegritätsfehler herbeiführen (Wells, 2001).

Abbildung 1 Deshalb betrügen Mitarbeiter
Zu Abbildung 1: Gelegenheiten entstehen, falls das Kontrollsystem Lücken aufweist und Handlungen dadurch unentdeckt bleiben können. Mitarbeiter glauben, dass sie nicht erwischt werden, dass es niemand herausfindet, dass die Konsequenzen nicht ernst sind und dass es somit das Risiko wert ist. Druck entsteht aus Angst vor Strafen für z.B. nicht erreichte Ziele, nicht eingehaltene Termine oder Vorgaben. Auch das Management kann Druck aufbauen, indem es entsprechende Signale (vermeintliche oder reale) sendet (wie unrealistische Abgabetermine oder Leistungskennzahlen, Budgetgrenzen, übermäßige Arbeitsbelastung oder Arbeitsrückstände). Rechtfertigung ist das Ergebnis von Einstellung und Ethik/Ehrlichkeit. Schlechtes Verhalten wird gerechtfertigt, um eine Handlung im Nachhinein als angemessen erscheinen zu lassen. Ein Beispiel: Man weiß zwar, dass eine bestimmte Handlung nicht richtig ist, weiß (oder glaubt) aber auch, dass es keine große Sache ist, da Andere dasselbe oder etwas Ähnliches gemacht haben. Rechtfertigung geht einher mit Ethik und Ehrlichkeit. Das Management sollte sich gewahr sein, dass nicht alle Beteiligten dieselbe Vorstellung davon haben, was eine ethische Handlungsweise ist. Was Ethik ist, liegt im Auge des Betrachters, und nicht jeder ist ehrlich.
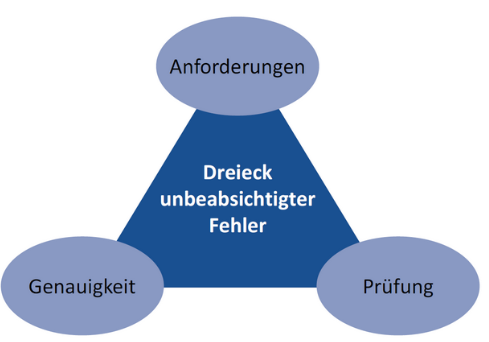
Abbildung 2 Gründe für die unbeabsichtigte Verletzung der Datenintegrität (Fehler)
Abbildung 2 gibt die Gründe für eine unbeabsichtigte Verletzung der Daten-integrität (z.B. Fehler) wieder.
In der realen Welt gilt: „Jeder macht Fehler, und Ethik begründet die Handlungen, die darauf folgen”. Mitarbeiter machen Fehler, weil sie vielleicht nicht über die Anforderungen informiert worden sind oder nicht verstanden haben, wie wichtig es ist, den vorgegebenen Anforderungen zu entsprechen. Auch sind die Dokumente, die die Anforderungen definieren, möglicherweise nicht klar und präzise genug verfasst und die Mitarbeiter ziehen eigene Schlüsse. Sind die Anforderungen unklar, können die Handlungen von Mitarbeitern abweichen von dem, was ursprünglich beabsichtigt war. Das wiederum kann Handlungen/Verhaltensweisen bedingen, die zu Fehlern führen, ohne dass der Mitarbeiter das realisiert. Genauigkeit bedeutet, dass die Aufzeichnungen widerspiegeln, was genau passiert ist. Mitarbeiter, die Daten aufzeichnen, mögen an der einen oder anderen Stelle einen Fehler machen, sei es aufgrund von Fahrlässigkeit, Ablenkung, Informationsdefiziten oder aus anderen Gründen.
Sind keine Systeme oder Kontrollen installiert, die die Korrektheit aufgezeichneter Informationen und Daten prüfen, können unbeabsichtigte Fehler unentdeckt bleiben. Unbeabsichtigte Fehler sind solche, bei denen der Mitarbeiter, der Daten und Informationen eingibt, nicht realisiert, dass er einen Fehler gemacht hat (z. B. nicht die Absicht hat, Daten oder Informationen in der Weise einzugeben, dass Aufzeichnungen oder Berichte nicht mehr genau wiedergeben, was tatsächlich passiert ist, oder z.B. das Auslassen relevanter Fakten). Da unbeabsichtigte Fehler an jeder Stelle des Sammelns und Berichtens von Daten und Informationen passieren können, muss die Korrektheit von einer zweiten Person (oder auf elektronischem Weg) geprüft werden. Dies ist wichtig, um sicherstellen zu können, dass Fehler entdeckt werden. Solange keine wirksamen Schritte ergriffen werden, Daten und Informationen genau auf ihre Korrektheit, Wahrheit und Vollständigkeit zu überprüfen, können unbeabsichtigte Fehler unentdeckt bleiben. Im Folgenden sind einige Beispiele zu unbeabsichtigten Fehlern aufgeführt, die bei sorgfältiger Überprüfung gefunden werden können:
- Versehentliches Aufzeichnen der falschen Zeit oder des falschen Datums.
- Nichteingabe benötigter Daten (Vergessen).
- Aufzeichnung falscher Messwerte.
- Übertragen der falschen Werte.
- Anwendung falscher Spezifikationen oder Grenzwerte.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











