

Disruptive Technologien in der Herstellung von Arzneimitteln
8 Min. Lesezeit | von
Experten: Martin Düblin, Dr. Rainer Gnibl | Moderation: Barbara Peither | Protokoll: Nadja Schaubhut
Erschienen im LOGFILE Leitartikel 16/2020
Ein GMP-DIALOG der GMP-BERATER-Tage im Oktober 2019 drehte sich um das Thema „Disruptive Technologien in der Herstellung von Arzneimitteln“. Dabei stellten die Teilnehmenden Fragen rund um das Thema, welche die beiden Experten im Rahmen einer spannenden Diskussion beantworteten.
Während moderne Technologien wie KI, Machine Learning und das Internet of Things in anderen Branchen längst zum Standard gehören, werden sie in der Pharmaindustrie noch recht wenig verwendet. Was sind die Bedenken? Wo gibt es schon Lösungen? Wie sieht unsere Zukunft aus? Wie stehen die Behörden den neuen Technologien gegenüber? Wäre ein „Reset“ in der Gesetzgebung nötig, um diese neue Welt voranzubringen.
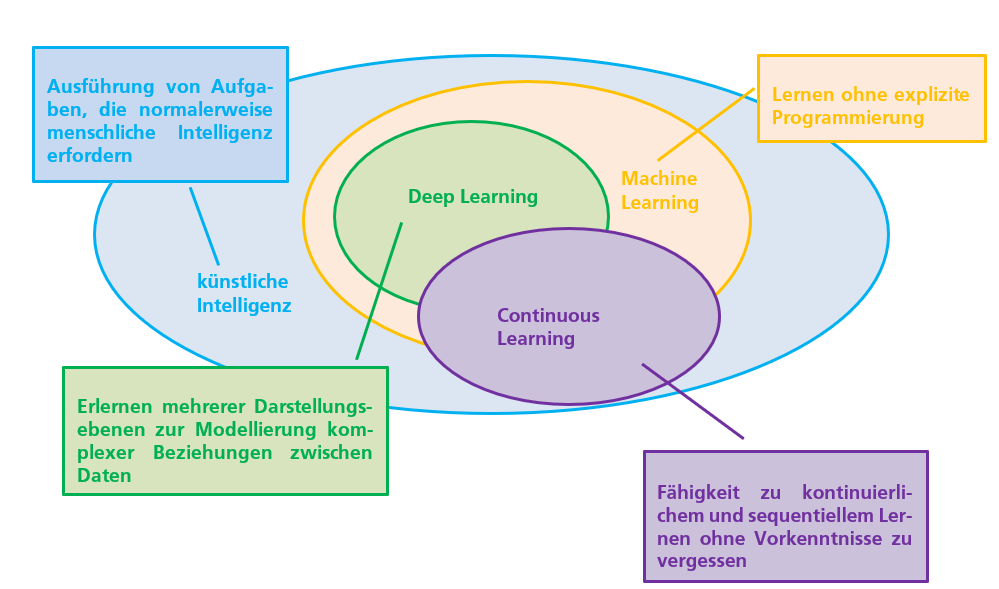
„Künstliche Intelligenz“ als Überbegriff (Abbildung frei nach Diagram 1 in „Perspectives and Good Practices for AI and Continuously Learning Systems in Healthcare“, Xavier University, Xavier Health Organization August 2018)
Kann man durch den Einsatz von Robotik auf das Vier-Augen-Prinzip verzichten? Wie kommt die KI „an die Spitze“ bzw. zu den Behörden und wie sieht es mit der behördlichen Akzeptanz aus?
Die erste Frage muss eigentlich gar nicht mehr gestellt werden: Das Vier-Augen-Prinzip kann jetzt schon abgedeckt werden. Es handelt sich ja dabei um ein Prinzip, dass die Fehlerrate von Menschen verringern soll. Der Algorithmus kontrolliert sich selbst. Ein System, das über „Machine Learning“ verfügt requalifiziert sich laufend selbst. Die Qualifizierung wird automatisch bestätigt und die Fehlerrate nimmt ab. Dazu ist keine Qualifizierungsphase mehr nötig. Darauf steuern wir in etwa fünf bis zehn Jahren zu. Für die Umsetzung müssen jedoch die Regularien angepasst werden. Bereits jetzt ist dieser Prozess zu langsam. Ein „Reset“ müsste her: Abstimmungsprozesse müssen beschleunigt werden. Dazu muss jedoch erst einmal ein Bewusstsein für das rasante Voranschreiten der Entwicklungen in diesem Bereich und damit für die Notwendigkeit von gesetzlichen Anpassungen geschaffen werden. Eine interdisziplinäre Zusammenarbeit ist dringend notwendig. Selbst innerhalb der Behörden ist die Zusammenarbeit nicht gegeben. Man muss über virtualisierte Umgebungen und deren Qualifizierung sprechen!
Damit kommt die Frage nach sogenannten „Robotergesetzen“ auf…
Bisher gibt es in Deutschland und auch Europa keine Rechtsgrundlage für eine solche Gesetzgebung. Als Beispiel dafür wurden autonom fahrende Autos genannt. Die Politik muss hier dringend handeln, denn die Welt ist bereit dafür. Im Vergleich zu China ist Europa hilflos, da sowohl die Politik als auch die Behörden zu weit von der Thematik entfernt sind. In China ist der Prozess der Gesetzgebung schneller. Es gibt eine technologische Führung und darum wird auch in Technologien investiert.
Gibt es Ansätze für eine Kombination von Robotik und PAT (Prozessanalysetechnologie) und sind Analysen bei Vollautomatisierung noch bezahlbar?
Das jetzige Wissen und die Vorgehensweisen müssen in die neuen Systeme implementiert werden. Die Grundlagen sind geschaffen, da kann sich, so der Behördenvertreter, auch keine Behörde verweigern. Die Machbarkeit wird durch andere Branchen vorgelebt. Die Vorgaben und Inhalte bleiben gleich. Dazu müssen jedoch neue Prozesse angestoßen werden. Als Beispiel wurde die sterile Arzneimittelherstellung genannt: Die ganze Kette muss funktionieren. Es kann und muss rund um die Uhr produziert werden, da die Kosten sonst zu hoch würden. Eine hohe Chargenanzahl ist daher eine Grundvoraussetzung. Die Traceability wäre ebenfalls gegeben, denn die Dokumentation ist direkt inbegriffen. Die Diagnostik arbeitet schon lange so. Bei einem 24/7-Betrieb mit geringen Ausfallzeiten überwiegen die Vorteile langfristig gesehen deutlich. Die gesetzlichen Grundlagen hierfür sind nicht zuletzt über den Annex 17 des EU-GMP-Leitfadens geschaffen.
Wer trägt die Verantwortung für die Arzneimittel und deren Qualität? Wie bekomme ich den Prozess valide und wer bestätigt das?
Bisher sind das Pharmaunternehmen bzw. die Personen in den entsprechenden Funktionen für die Arzneimittelqualität verantwortlich. Mit dem Zustand der Vollautomatisierung ändert sich dies. Die zuliefernden Unternehmen, welche die Automaten stellen, tragen einen Großteil der Verantwortung. Die Qualität wird durch das System bestätigt. Wir müssen dieses System definieren und sind ganz klar gefordert. Damit wird die Ausbildung der QP und der Behörden um einiges IT-lastiger und umfangreicher. Ein Umdenken muss stattfinden: Bisher kann nur das Ergebnis überprüft werden. Somit ist allein ein indirekter Nachweis durch einen Funktionstest möglich. Um den Fehler in einer Softwareprogrammierung zu finden, muss der Code mühsam durchsucht werden. Daher muss die Programmierung schneller und leichter überprüfbar werden. Es muss beim Programmieren der Software von Anfang an klar sein, was am Ende herauskommen soll.
Wird die Planung auch von Computern übernommen, wenn keiner mehr arbeitet?
Die Lösungen sind bereits auf dem Markt. Die Planung wird vom System übernommen.
Wie steht es um den Schutz der Daten vor Hackern? Kann man den Daten vertrauen? Wer kontrolliert die KI?
Ein Restrisiko ist nie auszuschließen. Kriminelle Energie kann immer vorhanden sein. Es ist bereits jetzt möglich, durch einen Hackerangriff die gesamte Produktion lahmzulegen. Lösungen werden nach und nach kommen.
Was passiert mit der freien Kapazität der Menschen, wenn sie nicht mehr arbeiten? Wie steht es um die Individualität, wenn alles auf Basis der gleichen Algorithmen funktioniert?
Im Jahr 2050/60 könnte alles vorhandene Wissen jederzeit allen zur Verfügung stehen. Somit entstünden gleiche Bildungschancen für alle. Das Leben im Rahmen eines akzeptablen Fußabdruckes wäre für alle möglich. Die Individualität wird damit nicht ausgeschlossen: Wer eine Tätigkeit gerne tut, kann diese nach wie vor ausüben. Zudem zeigt sich mit Blick auf die Zeitgeschichte: Trends kommen wieder. Vieles war schon einmal da und begegnet uns erneut im „Retro-Style“.
Wie kann man den Menschen die Angst vor neuen Technologien nehmen und warum sehen Roboter immer aus wie Menschen oder Tiere?
Die Menschen müssen miteinbezogen werden. Es ist nach wie vor mehr Aufklärung nötig, oft wird das Thema negativ dargestellt und Ängste werden geschürt. Jeder und jede Einzelne kann dazu beitragen, Informationen zu verbreiten. Roboter sind in ihrem Aussehen deshalb Menschen oder Tieren nachempfunden, weil sie in Tests dann besser akzeptiert wurden als beispielsweise ein Kasten in geometrischer Form. Zudem ersetzten Roboter in erster Linie menschliche Tätigkeiten.
Fazit:
- Die Lösungen sind größtenteils vorhanden, die regulatorische Anpassung hinkt jedoch hinterher.
- Die Risiken betreffen uns ebenfalls jetzt schon.
- Die interdisziplinäre und behördeninterne Zusammenarbeit muss dringend verbessert werden. Der gesamte Prozess muss überdacht werden.
- Die Öffentlichkeit muss stärker einbezogen werden, auch um Unsicherheiten aus dem Weg zu räumen.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











