

Dokumentenstruktur der Qualifizierung
Auszug aus dem GMP:KnowHow Anlagenqualifizierung
7 Min. Lesezeit | von Thomas Peither
Erschienen im LOGFILE 13/2024
Welche Dokumentenstruktur eignet sich für Ihre Qualifizierung? Phasenspezifisch oder phasenübergreifend? Kompakt oder modular?
Bei der Qualifizierung fällt eine große Zahl an Dokumenten an. Um hier den Überblick zu behalten, ist es wichtig, eine gut durchdachte und konsistente Struktur einzuführen. Eine regulatorisch definierte Dokumentenstruktur bei Qualifizierungsprojekten gibt es nicht. Das weiß GMP-Experte Thomas Peither. Er erläutert, was es bei der Dokumentenerstellung und -verwaltung zu beachten gibt.
Der Leitartikel ist ein Auszug aus dem GMP:KnowHow Anlagenqualifizierung, dem großen Lern- und Wissensportal speziell für die Qualifizierung in der Pharmaindustrie, das Sie kostenlos testen können.
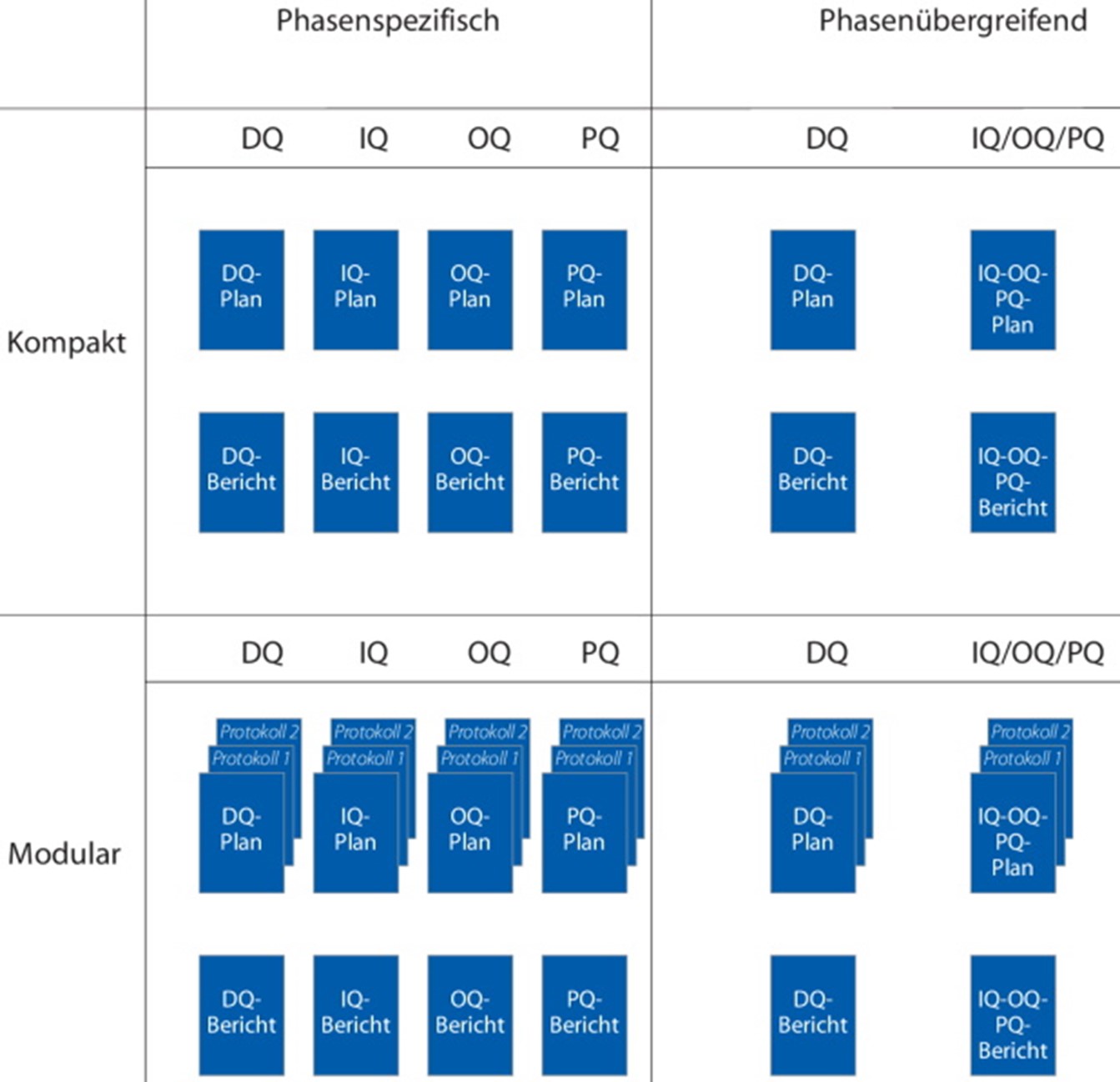
Phasenspezifisch oder phasenübergreifend? Kompakt oder modular? Welche Dokumentenstruktur eignet sich wann?
Bei der Qualifizierung fällt eine große Zahl an Dokumenten an. Um hier den Überblick zu behalten, ist es wichtig, eine gut durchdachte und konsistente Systematik einzuführen. Eine regulatorisch definierte Dokumentenstruktur bei Qualifizierungsprojekten gibt es nicht. Jedes Unternehmen hat daher Gestaltungsräume, seine eigene Dokumentenstruktur aufzubauen. Im Folgenden werden verschiedene Möglichkeiten aufgezeigt.
Die Dokumentenstruktur kann aus dem Qualifizierungsmasterplan (QMP) beschrieben werden oder Inhalt einer SOP sein, auf die im QMP referenziert wird. Welche Alternativen im Unternehmen Anwendung finden, ist sehr stark von dessen bestehenden Dokumentationsstrukturen abhängig. Prinzipiell gilt: Die modularen Konzepte eignen sich besser für eine Standardisierung als die kompakten Konzepte, denn die geeigneten Module für das jeweils zu qualifizierende System können damit einfach zusammengestellt werden. Außerdem lassen sich die notwendigen Anpassungen effizienter durchführen.
Die Designqualifizierung (DQ) und die Leistungsqualifizierung (PQ) bleiben in der Regel in allen Strukturen eigene Qualifizierungsphasen. Dies liegt daran, dass die DQ zeitlich klar abgegrenzt und häufig durch separate SOPs, z. B. durch den Einsatz von Risikoanalysen in der DQ, geregelt ist. Die PQ findet meist mit anderen technischen Systemen gemeinsam statt und hat daher fast immer eine andere Ausrichtung als DQ, IQ (Installationsqualifizierung) und OQ (Funktionsqualifizierung).
Bei der phasenspezifisch kompakten Vorgehensweise werden für jede Qualifizierungsphase Dokumente erstellt, die jeweils Prüfprotokolle mit dem zugehörigen Qualifizierungsplan zusammenfassen. Die Kombination von Beschreibungen der Prüfpunkte in Qualifizierungsplänen mit der Dokumentation der durchgeführten Prüfungen in den Qualifizierungsprotokollen hat folgende Vorteile:
- Nur ein Dokument muss verwaltet und gepflegt werden.
- Jede Phase wird separat durchgeführt und abgeschlossen.
In der Praxis hat sich darüber hinaus eine Sonderform dieser Dokumentenstruktur entwickelt, bei der der Bericht in den Plan integriert wird. Nach Freigabe des Plans werden die Prüfungen im selben Dokument protokolliert, bewertet und erlangen mit der zweiten Freigabe den Charakter eines Berichtes. Mit dieser komprimierten Form gelingt es, in einem Dokument zur xQ-Phase die geplante Vorgehensweise zu beschreiben und die Ergebnisse zu dokumentieren. Das reduziert die Gesamtzahl der zu verwaltenden Dokumente und somit auch den Archivplatz, bedarf aber einer intensiven Vorbereitung.
Unregelmäßigkeiten im zeitlichen Verlauf der Dokumentenerstellung sind leicht zu entdecken, da alle freigaberelevanten Datumsangaben in einem Dokument vorliegen.
Die phasenspezifische modulare Qualifizierungsstruktur verfügt über Qualifizierungspläne, die so aufgebaut sind, dass die generelle Struktur der Prüfprotokolle einmal festgelegt wird. Diese Prüfpläne werden dann in den einzelnen Qualifizierungsphasen lediglich angepasst und dem Qualifizierungsplan beigefügt.
Diese Systematik hat den Vorteil, dass die Verwaltung und Pflege der Qualifizierungspläne getrennt von den Prüfprotokollen erfolgt. Damit ist auch eine zeitlich versetzte Erstellung möglich, ggf. auch mit unterschiedlichen Genehmigungsverfahren. Der Nachteil ist, dass die Anzahl der Dokumente deutlich größer ist als bei den bisher vorgestellten Varianten.
Im Vergleich zur phasenspezifischen werden bei der phasenübergreifenden Struktur mehrere Qualifizierungsphasen in einem Dokument zusammengefasst. Häufig findet diese Variante Anwendung bei der Kombination von IQ und OQ. Bei diesem Modell gilt es, sicherzustellen, dass die Prüfungen der einzelnen Phasen zeitlich nacheinander erfolgen. Zwischen den beiden Phasen gibt es eine Zwischenfreigabe. Vor Beginn der nächsten Qualifizierungsphase müssen alle offenen kritischen Punkte abgearbeitet sein. Für noch offene Punkte ist zu begründen, warum diese für die Weiterführung der Qualifizierung nicht kritisch sind.
Diese Struktur reduziert die Anzahl von Genehmigungsdokumenten erheblich und kann die Projektbearbeitung beschleunigen. Insbesondere muss nach der IQ zunächst kein formeller IQ-Bericht erstellt werden, bevor die OQ-Arbeiten starten können. Stattdessen geben die Verantwortlichen die Freigabe zur Durchführung der OQ. Diese Zwischenfreigabe bildet somit formell den Abschluss der IQ-Arbeiten. Auch ist ein solcher IQ-Abschluss weniger aufwendig als ein kompletter IQ-Bericht. Dennoch trägt der dokumentierte Abschluss den Charakter eines offiziellen Dokumentes und fließt mit dem OQ-Bericht in die Gesamtdokumentation ein.
Nachteilig ist, dass die Phasen untereinander nicht sauber abzugrenzen sind. Es müssen alle Informationen bei der Erstellung der xQ-Pläne und Prüfprotokolle vorhanden sein, was insbesondere bei komplexen Qualifizierungsprojekten nicht immer gewährleistet ist. Die DQ und die PQ werden, wie schon beschrieben, in der Regel separat dokumentiert.
Im phasenübergreifenden modularen Konzept werden in der Regel der IQ-/OQ- und PQ-Plan zusammen erstellt und die zugehörigen Prüfprotokolle als separate Dokumente geführt. Auch bei diesem Modell ist sicherzustellen, dass die Prüfungen der verschiedenen Qualifizierungsphasen zeitlich nacheinander abgearbeitet werden können. Wie bereits ausgeführt, folgt zur Abgrenzung der einzelnen Phasen in der Regel eine Zwischenfreigabe nach der IQ bzw. OQ.
Diese Struktur hat den Vorteil, dass die Anzahl der Dokumente reduziert und administrative Informationen gebündelt werden. Obwohl IQ-, OQ- und PQ-Plan zusammengefasst sind, bleibt die Modularität durch die separaten Protokolle erhalten. Die xQ-Prüfprotokolle können separat verwaltet und gepflegt werden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











