

eDMS/eQMS: Aufbau, Anwendung und GxP-Anforderungen
Auszug aus dem GMP-BERATER, Kapitel 1.H.2, Aufbau elektronischer Dokumentenmanagement- und Qualitätsmanagement-Systeme (eDMS/eQMS)
7 Min. Lesezeit | von Thilo Gukelberger
Erschienen im LOGFILE Leitartikel 46/2021
Bei der Auswahl eines geeigneten eDMS/eQMS ist darauf zu achten, dass die vorgenannten Autorisierungsprüfungen immer durchlaufen werden, egal, mit welcher Bedienoberfläche der Anwender mit dem eDMS/eQMS kommuniziert.
Ein eDMS speichert im Wesentlichen folgende elementare Informationsobjekte:
- die Dokumente selbst (z. B. Word-, Excel-, PDF-Dateien)
- Attribute/Metadaten der Dokumente (z. B. Titel, SOP-Nummer, Autor, Chargennummer, Sachgebiet)
- Beziehungen zwischen Anwendern und Dokumenten (Rechte, Rollen, Organisationsstruktur)
Die Dokumente selbst werden entweder in schreibgeschützten Speicherbereichen (Storagesysteme mit sogenannter Soft-Worm-Funktionalität) als Datei oder aber in einer relationalen (SQL-) Datenbank als sogenannte BLOBs (Binary Large Objects) abgelegt.
Die Attribute/Metadaten ermöglichen eine automatisierte Gruppierung bzw. Bündelung derjenigen Dokumente, die in inhaltlichem Zusammenhang stehen. Man spricht dann auch von einer Akten- bzw. Dossierbildung. Die Attribute/Metadaten werden fast immer in einer Datenbank (zumeist relationale SQL-Datenbank) abgelegt.
Beispielhaft für die Bündelung von Dokumenten sei hier das elektronische Chargenprotokoll (electronic batch record) erwähnt. Betrachtet man die Chargennummer als gemeinsames Attribut, das für alle Dokumente eines Chargenprotokolls (z. B. Herstellprotokoll, Prüfprotokoll, Verpackungsprotokoll) identisch ist, so kann ein eDMS jedes Dokument des Herstellprozesses bei der elektronischen Ablage automatisch in die korrekte Chargenakte einfügen.
Den Vorgang des Einfügens eines Dokumentes in eine Akte/Dossier darf man sich an dieser Stelle nicht wie bei der papierbasierten Dokumentation vorstellen. Papierdokumente können immer nur an einem physischen Speicherort abgelegt werden. Benötigt man das Dokument an unterschiedlichen Orten, so muss es entweder manuell transportiert oder kopiert werden. Beide Alternativen haben große Nachteile: Ein manueller Transport birgt die Gefahr von Dokument‑, auf jeden Fall aber Zeitverlusten, eine Papierkopie die Gefahr von Inkonsistenzen zwischen Original und Kopie. Beim Erstellen einer Akte oder eines Dossiers speichert ein eDMS in der Regel nur einen Link auf das einzufügende Dokument. Das eDMS führt also nur eine einzige Instanz des Dokumentes und präsentiert dieses den Anwendern in beliebigen prozessualen Kontexten.
Der Arbeitsablauf mit einem eDMS/eQMS stellt sich üblicherweise wie in Abbildung 1.H-1 gezeigt dar.
 Abbildung 1.H-1 Arbeit mit einem eDMS / eQMS (Schema)
Abbildung 1.H-1 Arbeit mit einem eDMS / eQMS (Schema)
- Start: Der Anwender startet seine eDMS/eQMS-Anwendung. Dies kann entweder eine spezielle Anwendung (nativer Client) oder ein Webbrowser sein.
- Authentisierung: Der Anwender authentisiert sich am eDMS/eQMS. Nutzt man hier Mechanismen wie „single sign on“, so ist man durch seine Anmeldung im Firmennetzwerk bereits automatisch authentifiziert. Im GxP-Umfeld muss sich der Anwender jedoch spätestens beim Leisten seiner elektronischen Unterschrift erneut authentifizieren.
- Suche: Der Anwender sucht nach Dokumenten. Dazu werden durch Eingabe in der Bedieneroberfläche des Systems Suchkriterien an den eDMS/eQMS-Server geschickt.
- Autorisierung: In Abhängigkeit der Zugriffsrechte des angemeldeten Benutzers liefert der eDMS/eQMS-Server eine adäquate Trefferliste. Diese besteht zunächst nur aus den Metadaten der gelieferten Dokumente.
- Funktion: Der Anwender wählt nun den gewünschten Treffer aus und bestimmt – meist über das Kontextmenü seiner Bedienoberfläche – welche Funktion er mit dem ausgewählten Dokument ausführen möchte (z. B. „Dokument anzeigen“, „Dokument überarbeiten“, „Dokument prüfen“).
- Autorisierung: Der eDMS/eQMS-Server überprüft, ob der Anwender die notwendigen Rechte besitzt und führt gegebenenfalls die gewünschte Aktion mit dem Dokument durch.
Bei der Auswahl eines geeigneten eDMS/eQMS ist darauf zu achten, dass die vorgenannten Autorisierungsprüfungen immer durchlaufen werden, egal, mit welcher Bedienoberfläche der Anwender mit dem eDMS/eQMS kommuniziert.
Ein eDMS, das auch außerhalb des GxP-Bereiches Anwendung findet, beinhaltet meist Funktionen, die nicht GxP-konform sind. Hat man in diesem Fall Zusatzmodule, die die Handhabung GxP-relevanter Dokumente sicherstellen, so muss gewährleistet sein, dass diese Dokumente auch nur mittels dieser Zusatzmodule zugänglich gemacht werden.
Abbildung 1.H-2 zeigt Funktionen, die im GxP-Umfeld unerlässlich sind, oft aber bei eDMS-Systemen fehlen:
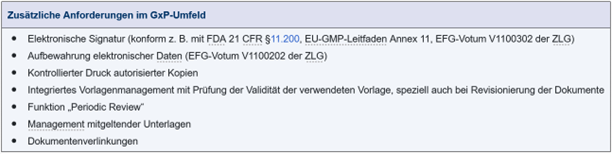
Abbildung 1.H-2 Zusätzliche Anforderungen im GxP-Umfeld
Abgesehen vom funktionalen Umfang einer eDMS/eQMS-Lösung muss diese natürlich validierbar sein. Hierzu bedarf es guter Kenntnisse der Systemarchitektur und der Systemfunktionen. Idealerweise liefert der Hersteller ein prevalidiertes System, so dass der Aufwand der projektspezifischen Validierung minimiert werden kann. Ein prevalidiertes System sollte zumindest die Dokumente für die IQ, OQ, DQ beinhalten, ggf. als Vorlagen, die projektspezifisch angepasst werden müssen.
Weitere Hinweise zur Funktionsweise von Dokumentenmanagementsystemen und zur Auswahl eines geeigneten Systems für den GxP gerechten Betrieb finden Sie in Kapitel 15.G Dokumentenmanagementsysteme.
Im Folgenden wollen wir den Fokus weg von der reinen Sicht auf Dokumente und hin zum übergeordneten Blick auf die Prozesse lenken.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











