

Erzeugung von WFI mit Membrantechnik
Ein Auszug aus dem GMP-BERATER, Kapitel 5.B.11, Erzeugung von WFI mit Membrantechnik
8 Min. Lesezeit | von Dr. Herbert Bendlin und Fritz Röder
Erschienen im LOGFILE Leitartikel 22/2021
Lange Zeit war in Europa zur Erzeugung von Wasser für Injektionszwecke (WFI) die Verdampfung des Wassers (z. B. Destillation) zwingend vorgeschrieben. Dies hat sich im April 2017 geändert:
seit diesem Zeitpunkt sind auch Membranverfahren zugelassen. Diese wurden bisher zur Erzeugung von Hochgereinigtem Wasser (HPW) eingesetzt. Diese Wasserqualität, die sich bis dato nur durch die Herstellungsmethode von WFI unterschied, ist seit April 2019 nicht mehr im Europäischen Arzneibuch enthalten. Die Erzeugung von WFI mittels Membrantechnik wird auch als „kalte“ WFI-Herstellung bezeichnet. Derzeit wird der Vergleich zwischen den „heißen“ Verfahren (Destillation und Thermokompression) und den „kalten“ Verfahren (Membrantechnik) in der Industrie und bei den Behörden intensiv diskutiert. Zu bedenken ist in diesem Zusammenhang, dass die Anwendungsrisiken für Arzneimittel, für die man WFI verwendet, höher sind als für Arzneimittel, die bisher mit HPW hergestellt wurden, beispielsweise Ophthalmika (Produkte zur Anwendung am Auge). Arzneimittel, die parenteral verabreicht werden und WFI enthalten, verteilen sich direkt im Blutkreislauf. Es gibt keinerlei Barriere mehr, die eventuelle Verunreinigungen zurückhalten könnte. Allein durch die Anwendung des Arzneimittels (Applikationsweg) ergeben sich also höhere Risiken und somit Anforderungen an die Technik. Eine Qualifizierung von WFI-Membrananlagen muss daher mit entsprechender Sorgfalt durchgeführt werden. Die Risiken der kalten Herstellung werden seitens der Hersteller, Betreiber und Behörden noch unterschiedlich beurteilt. Auch spielt die Betriebsweise und Überwachung beim Anwender eine größere Rolle als sie bei der rein technischen Bewertung zum Tragen kommt.
Wenn man auf Membrantechnik zur WFI-Erzeugung umstellen möchte, ist die zuständige Überwachungsbehörde darüber zu informieren. Dies ist eine Anforderung der EP-Monographie 0169.
Der wesentliche Vorteil der Membrantechnik liegt in den geringeren Kosten: bisher hatte man ein System für Gereinigtes Wasser und schaltete eine zweite Anlage zur Verdampfung nach, um WFI zu erhalten. Man brauchte auch zwei Lager- und Verteilsysteme. Das alles kann nun entfallen. Man stellt nur noch WFI per Membranverfahren her und benutzt dieses für alle Anwendungen. So entfallen die Anschaffungs-, Betriebs- und Energiekosten für das heiße System sowie die zweite Verteilung im Gebäude.
Der Aufbau einer Anlage zur Herstellung von HPW-Wasser oder kaltem WFI ist in Abbildung 5.B-32 dargestellt.
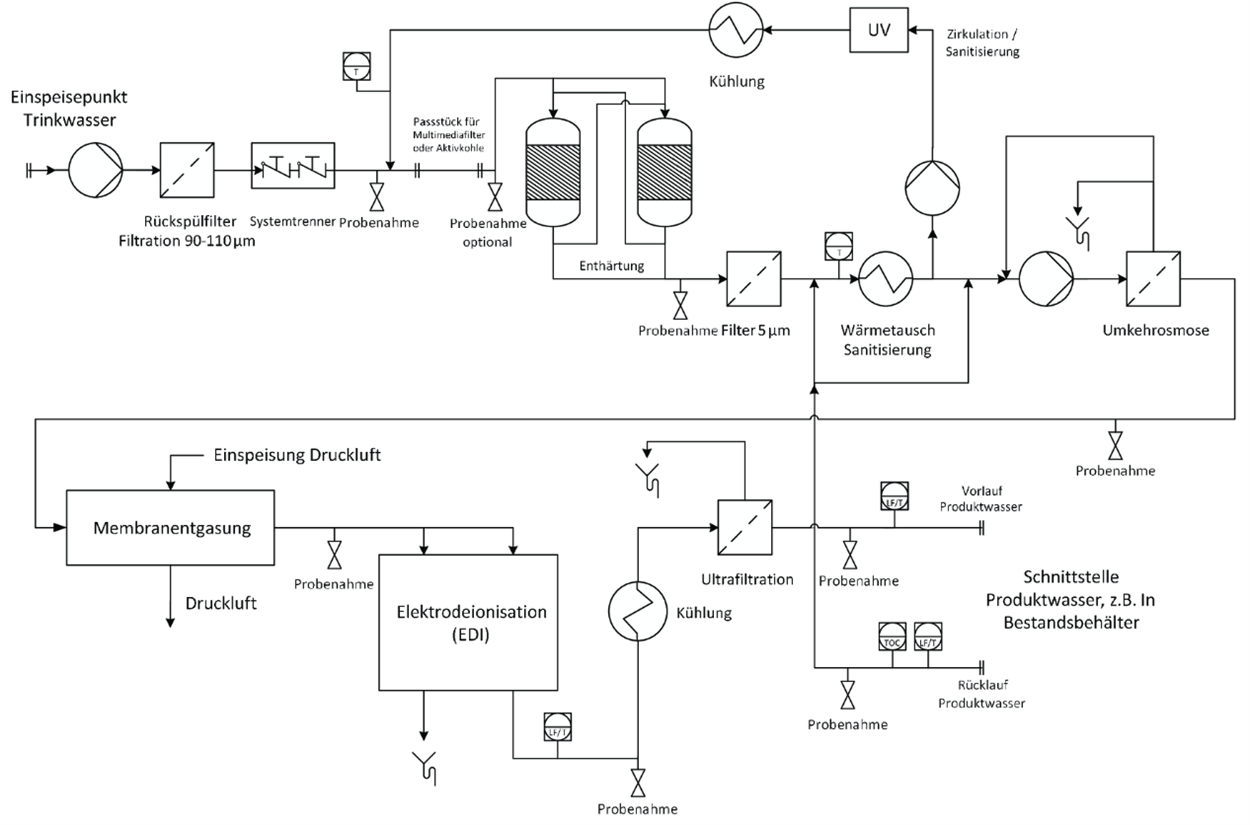
Abbildung 5.B-32 Schema einer Anlage zur Herstellung von HPW oder kaltem WFI
Aufgrund der erhöhten Risiken bei der parenteralen Anwendung weist die aktuell gültige Leitlinie1 der EMA auf verschiedene zusätzliche Kontrolleinrichtungen hin, die bis dato bei HPW nicht notwendige Praxis waren. Darunter fallen zum Beispiel:
- Der Einsatz von Schnellmethoden für Keimzahl- und Endotoxinbestimmung (in Verbindung mit den konventionellen Bestimmungsmethoden gemäß Monographie)
- Der Einsatz kombinierter Sanitisierungsverfahren mittels Hitze UND Chemikalien (siehe Kapitel 5.E.7.1 Sanitisierung)
- Erhöhter Wartungs- und Überwachungsaufwand (Austausch von RO-Membranen/Beprobungsfrequenzen)
- Der Einsatz von TOC-Messtechnik an mehreren Stellen im System (siehe Kapitel 5.C.6.7 TOC-Messung (online))
Weitere geforderte Maßnahmen, die aber auch für HPW bereits galten, sind z. B.:
- Nachweis der Integrität der UF-Module
- Analyse der Rohdaten mittels statistischer Tools zur Bewertung der Warn- und Aktionsgrenzen
- Eine ganzheitliche Prozesskontrollstrategie
Erste Anlagen zur WFI-Herstellung mit Umkehrosmose sind bereits in Betrieb gegangen (Stand: Mai 2019). Die Reaktionen der Behörden in Europa dazu fielen bisher sehr unterschiedlich aus (von „keine Reaktion“ bis zu „Vor-Ort-Besuch“).
Schnellmethoden zur Anlagenüberwachung
Die sogenannten Schnellmethoden (rapid microbiological methods) betreffen die Bestimmung von Keimzahl, TOC-Wert und Endotoxinen im Wasser. Alle drei werden im Q&A-Dokument der EMA andiskutiert. Der wesentliche Vorteil dieser Bestimmungsverfahren liegt darin, dass das Ergebnis wesentlich früher vorliegt und so Risiken vermieden werden.
Die Online-Bestimmung des TOC-Wertes ist bereits gängige Praxis. Es werden nur deutlich mehr Stellen zur Überwachung empfohlen als bisher.
Was die Online-Keimzahlbestimmung angeht, so sind derzeit noch nicht viele Geräte auf dem Markt. Erste Erfahrungen der Hersteller haben noch Probleme mit der Messtechnik aufgezeigt. Es ist zu hoffen, dass die Messtechnik in naher Zukunft so optimiert wird, dass ein Umgang mit den Messdaten in der Praxis gut möglich wird. Zusätzlich muss man sich darüber im Klaren sein, dass eine Online-Messung definitiv andere Ergebnisse liefert als die klassische Beprobung. Die Messergebnisse einer Online-Keimzahlmessung und einer klassischen Inkubation korrelieren nicht miteinander. Die Wasserfreigabe erfolgt gemäß der gängigen Monographien immer noch auf Basis der Offline-Daten. Die Online-Bestimmung der Keimzahl ist daher ausschließlich als Teil der Prozesskontrollstrategie zu betrachten und kann nur unterstützend und zur Fehlersuche eingesetzt werden.
Auch für Endotoxine sind erste Schnellmethoden verfügbar. Hier handelt es sich derzeit allerdings immer noch um Offline-Bestimmungsgeräte. Da ein LAL-Test schneller (ca. 1 Tag) als eine klassische Keimzahlbestimmung (ca. 5 Tage) durchgeführt werden kann, ist die Zeitersparnis bei den Endotoxin-Schnellmethoden nicht so groß wie bei der Keimzahlbestimmung mittels einer Schnellmethode. Die Endotoxin-Schnellmethoden sind noch neu auf dem Markt. Erste Versuche haben auch hier Probleme bei der Methodenvalidierung gezeigt.
Insgesamt ist die Technik der Schnellmethoden sehr zu begrüßen, da sie das Maß an Prozesskontrolle deutlich verbessert. Allerdings bildet die Messtechnik die bisherigen Methoden noch nicht eindeutig nach und birgt noch Probleme bei der Methodenvalidierung. Es bleibt zu hoffen, dass die Technik weiter verbessert und optimiert wird und die Anlagen in Zukunft noch besser und einfacher überwacht werden können. Davon dürften nicht nur die Betreiber, sondern auch die Patienten profitieren.
Umgang mit bestehenden HPW-Anlagen
Seit April 2019 gibt es im Europäischen Arzneibuch keine Monographie mehr für HPW. Auch die „Note for Guidance“ der EMA zu Wasser (Kapitel G.4) wird derzeit überarbeitet, HPW wird hier ebenfalls entfallen. In der Praxis sind allerdings noch viele HPW-Anlagen in Betrieb. Viele Nutzer fragen sich daher, was nun ohne eine gültige Monographie zu tun ist. Nachfolgend wird ein nach Ansicht des Autors möglicher Ansatz zum Umgang mit HPW-Bestandssystemen vorgestellt:
Prüfen Sie Ihre Registrierungsunterlagen: Sind die Arzneimittel, die Sie herstellen, mit HPW registriert?
- Wenn nein, sind keine weiteren Aktionen erforderlich.
- Wenn ja, prüfen Sie, ob eine ältere Version des Europäischen Arzneibuchs vor dem Supplement 9.7 erwähnt ist.
- Ist das der Fall, können Sie sich auf die ältere Version des Arzneibuchs beziehen. Dokumentieren Sie den Sachverhalt entsprechend. Allerdings könnten Neuzulassungen Sie dazu bringen, Ihr System doch auf WFI umzurüsten.
- Wenn kein Bezug zu einer älteren Arzneibuch-Ausgabe besteht, ist es ratsam, diesen Umstand mit der zuständigen Überwachungsbehörde zu diskutieren.
Falls Sie Ihr HPW-System zu einem WFI-System umrüsten möchten, sind folgende Dinge zu beachten:
- Erstellen Sie eine Prozesskontrollstrategie, sofern noch nicht vorhanden.
- Die Überwachung und Kontrolle des Systems muss erhöht werden. Gute Hinweise hierzu finden Sie in dem neuen ISPE-Handbuch zu WFI mit Membrantechnik.
- Der Wartungsaufwand erhöht sich gegebenenfalls.
- Zusätzliche Messtechnik wird vermutlich notwendig.
- Eine Änderungsqualifizierung wird notwendig
Dieser Text ist ein Auszug aus dem GMP-BERATER, Kapitel 5.B.11
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











