

GMP/GDP-Anforderungen an unterschiedliche Lagerbereiche
Ein gekürzter Auszug aus der 34. GMP & TEA-Episode „Lagerung von Arzneimitteln“
8 Min. Lesezeit | von Thomas Peither
Erschienen im LOGFILE Leitartikel 22/2023
Die Lagerung von Material und Ware in der Arzneimittelherstellung birgt viele Risiken. Nicht ohne Grund steht sie häufig im Fokus von Inspektionen. Substanzveränderungen, Kontaminationen oder Verwechslungen können schwerwiegende Folgen haben. Mit den richtigen Maßnahmen können Sie diese Risiken aber minimieren und die Qualität Ihrer eingelagerten Ware sicherstellen.
- Wie sind die Verantwortlichkeiten geregelt?
- Welche Anforderungen gelten für unterschiedliche Lagerbereiche?
- Und: Welche Prüfungen sind beim Wareneingang erforderlich?
Die Antworten auf diese Fragen, Infos zu den geltenden Regularien und wertvolle Praxistipps für die direkte Umsetzung – all das erhalten Sie in der neuen Episode des Video-Podcasts GMP & TEA: „Lagerung von Arzneimitteln“. Lesen Sie hier direkt einen Auszug aus dem spannenden Webcast.
Allgemeine Anforderungen. – Eine ausreichende Größe und Beleuchtung aller Bereiche, in denen Arzneimittel sowie auch Ausgangsstoffe und Packmittel gelagert werden, ist für die ordnungsgemäße Durchführung der Arbeitsabläufe unerlässlich. Der Zugang zu den Lagerbereichen muss klar geregelt und geschützt sein.

Sowohl der EU-GMP-Leitfaden als auch die EU-GDP-Leitlinien betonen die Wichtigkeit einer vor Witterungseinflüssen geschützten Lagerung. Die Annahme von Waren sollte deshalb grundsätzlich innerhalb des Lagergebäudes erfolgen. Nur in Ausnahmefällen ist sie unter einem Vordach im Freien akzeptabel.
Zudem sollten die Bereiche für die Warenannahme und den Versand voneinander getrennt sein. Hat man wenig Platz, sind zusätzliche organisatorische Maßnahmen notwendig, zum Beispiel An- und Auslieferungsvorgänge zeitlich zu entkoppeln oder entsprechende Bereiche mindestens durch mobile Stellwände oder Absperrketten abzutrennen.
Gesonderte Lagerbereiche
Zurückgewiesene, gesperrte und aus dem Handel zurückgegebene Materialien sind als „gesperrt“ zu kennzeichnen und in einem separaten, nur autorisierten Personen zugänglichen Sperrlager aufzubewahren.
Gesonderte Lagerbereiche sind auch für hochaktive Stoffe vorgeschrieben. Nach der WHO-GSDP-Leitlinie (Good Storage and Distribution Practices for Medicinal Products) gilt dies auch für radioaktive Materialien, Betäubungsmittel, Stoffe mit Missbrauchsrisiko oder mit Brand- oder Explosionsgefahr, Gefahrstoffe oder empfindliche Stoffe.

Besondere Anforderungen an die Lagerung kritischer Materialien werden unter anderem in der Betäubungsmittelverordnung und Gefahrstoffverordnung definiert. Auch bedrucktes Verpackungsmaterial gilt unter GMP-Gesichtspunkten als kritisch; unter Umständen ist ein besonderes klimatisiertes Zwischenlager zur Konditionierung für den Verpackungsprozess nötig.
Eine gesonderte Lagerung ist für Stoffe mit abweichenden klimatischen Lagerungsbedingungen erforderlich. Häufig braucht es dafür spezielle Kühllager oder auch Lagerbereiche mit besonderen Spezifikationen für die relative Feuchtigkeit. Grundsätzliche Vorgaben finden sich hierzu sowohl im EU-GMP-Leitfaden als auch in den GDP-Leitlinien und im 21 CFR 211.
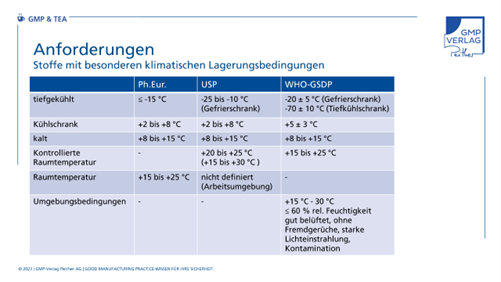
Auf Stabilitätsdaten basierte Lagerungshinweise gelten als Soll-Werte für Lagerungsbedingungen, um beispielsweise Haltbarkeitsdaten übernehmen zu können. Die auf der Packung des Fertigarzneimittels genannten Temperaturvorgaben zur Lagerung sind in Übereinstimmung mit der EMA-Guideline on Declaration of Storage Conditions zu wählen. In einschlägigen Arzneibüchern und der WHO-GSDP finden sich Lagerungshinweise für bestimmte Temperaturbereiche.
Aber Vorsicht! Begriffe wie „tiefgekühlt“ oder auch „Raumtemperatur“ können je nach Quelle unterschiedliche Temperaturen bedeuten. Die folgende Gegenüberstellung der Begriffe und Anforderungen in den verschiedenen Regelwerken kann hier hilfreich sein.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











