

GMP-konforme Anlagenqualifizierung: Softwarequalitätsplan
Ein Auszug aus dem GMP:KnowHow Anlagenqualifizierung, Kapitel „Softwarequalitätsplan“
7 Min. Lesezeit | von GMP-Verlag Peither AG
Erschienen im LOGFILE 15/2023
Ein Softwarequalitätsplan – was ist das, und wie erstellt man ein solches Dokument?
Kurz: In einem Softwarequalitätsplan wird beschrieben, wie eine Software geplant getestet werden soll.
Üblicherweise wird der Softwarequalitätsplan nach Erstellung einer Benutzeranforderung und einer Risikoanalyse zur entsprechenden Software angefertigt. Es wird geprüft, ob die erstellten risikobasierten Benutzeranforderungen in der Software umgesetzt werden.
Doch:
- Welche Informationen benötige ich dazu?
- Woher bekomme ich die Informationen?
- Wie sieht die Planung der Testphasen aus?
- Wie sieht die Struktur eines Softwarequalitätsplans aus?
Das sind die Leitfragen bei der Erstellung eines Softwarequalitätsplans.
Ein Softwarequalitätsplan und der Softwarequalitätsbericht werden von einer Person geschrieben, die sich nur mit der Erstellung des Plans bzw. Berichts befasst. Eine andere Person wird die Tests ausführen. Diese beiden Funktionen müssen getrennt sein. In einem Softwarequalitätsplan wird nicht nur davon ausgegangen, dass die Tests positiv ausfallen können („Software funktioniert wie gewünscht“). Auch ein mögliches negatives Ergebnis („Software funktioniert nicht wie gewünscht“) muss bei der Planung berücksichtigt werden.
Achtung: Der Softwarequalitätsplan basiert nicht auf Annahmen. Stattdessen müssen Akzeptanzkriterien definiert werden, welche für die erfolgreiche oder nicht erfolgreiche Testphase der Software ausschlaggebend sind.
Nachfolgend ein beispielhafter Aufbau, wie ein Softwarequalitätsplan aussehen könnte:
Am Anfang des Softwarequalitätsplans werden das Ziel und der Zweck des Dokuments beschrieben. Anschließend wird der Validierungsbereich definiert, d. h. für welches technische System der Softwarequalitätsplan gültig ist.
Beispiel: Der vorliegende Softwarequalitätsplan ist für die Software A123 mit den Schnittstellen XYZZ1 am Standort ABC gültig. Er soll dazu dienen, den Validierungsstatus für die Software herzustellen.
Außerdem werden kurz die Verantwortlichkeiten des Validierungsteams und erreichbare Fristen für die verschiedenen Phasen beschrieben.
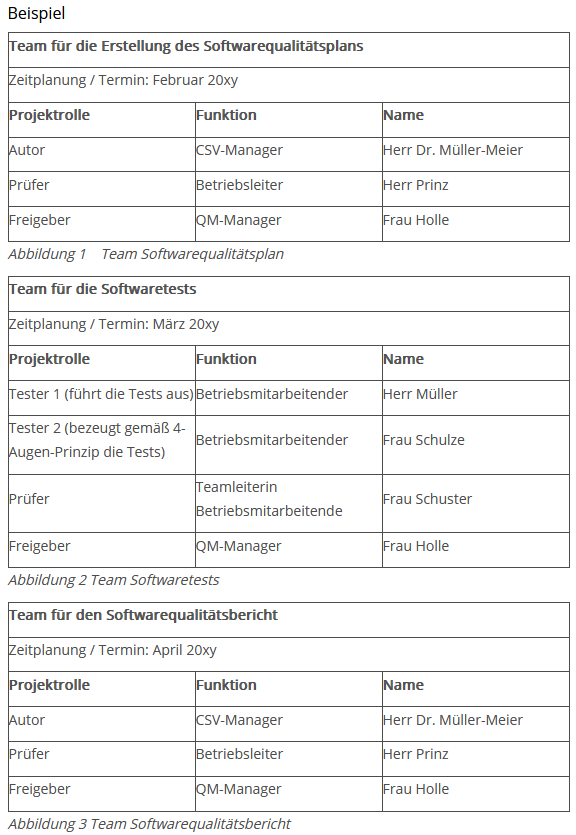
Die tabellarische Form ist übersichtlich und leicht verständlich.
Schulung
Die Personen, die an der Softwarequalitätstestung mitwirken, müssen für den Softwarequalitätsplan geschult werden. Das ist mit einem Schulungsnachweis zu dokumentieren.
Softwarebeschreibung / Computer–System-Beschreibung
Zu dem Softwarequalitätsplan gehört eine kurze Beschreibung folgender Aspekte:
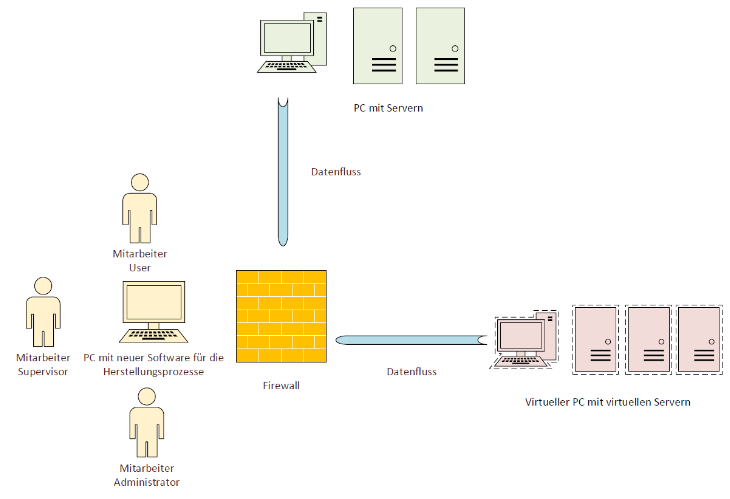
Abbildung 4 Software/Computer-System-Beschreibung
- Welche Software wurde beschafft (Name, Hersteller, Version)?
- Welche Funktion erfüllt die Software? Beispielsweise die Steuerung eines Herstellungsprozesses oder die Datenverwaltung eines Datenbanksystems?
- Wie ist die Software bzw. das Computer-System aufgebaut, und wie wird sie/es in bestehenden Systemen eingebunden?
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de










