

Grundlagen zur Dichtigkeit von Container Closure Systemen
Auszug aus dem GMP-BERATER, Kapitel 12.G.2, Grundlagen zur Dichtigkeit von Container Closure Systemen
5 Min. Lesezeit | von Raimund Brett
Erschienen im LOGFILE 03/2025
Unter dem Punkt „Finishing of sterile products“ sind im Annex 1 des EU-GMP-Leitfadens die Anforderungen zur Prüfung von sterilen Produkten beschrieben. Unter dem Finishing versteht man zum einen die Prüfung auf Dichtigkeit und zum anderen die Prüfung auf visuelle Verunreinigungen bzw. Partikel.
In unserem heutigen Leitartikel berichtet Raimund Brett über die Grundlagen zur Dichtigkeit von Primärverpackungssystemen (Container Closure System). Dabei geht es unter anderem darum, wie Undichtigkeiten definiert werden und welche Anforderungen an die Qualität von Container Closure Systemen gestellt werden.
Der Leitartikel ist ein Auzug aus dem GMP-BERATER, dem weltweit größten Standardwerk für Qualitätsmanagement in der Pharmaindustrie.
Die Aufgabe eines Primärverpackungssystem (Container Closure System) liegt im Wesentlichen darin, zu verhindern, dass das Produkt verloren geht (z. B. Auslaufen, Verdunsten etc.) und dass das Produkt von außen kontaminiert wird.
Eine Primärverpackung bzw. ein Verpackungssystem kann aus einem oder mehreren Bestandteilen bestehen. Als Beispiel seien hier Ampullen (nur eine Komponente, Glas) und Vials (mehrere Komponenten, Glasbehälter, Stopfen (Gummi) und Kappe (Aluminium)) genannt.
Folgende Definitionen können im Zusammenhang mit Undichtigkeiten gemacht werden:
- Leck/Undichtigkeit: Ein Spalt oder ein Bruch im Verpackungssystem worüber Flüssigkeit oder Gas austreten kann
- Leckage: Das unbeabsichtigte Eindringen oder Entweichen von Stoffen (fest, flüssig, gasförmig) durch eine Lücke in der Verpackung oder einen Spalt zwischen Verpackungsbestandteilen
Ein dichtes bzw. integres Verpackungssystem ist eines, das keine Undichtigkeiten wie Löcher oder Beschädigungen aufweist, die möglicherweise die Qualität des Produktes gefährden können.
Für Verpackungssysteme für sterile Produkte könnte man die Undichtigkeiten gemäß Abbildung 1 zum Beispiel in drei Kategorien einteilen (angelehnt and die USP).
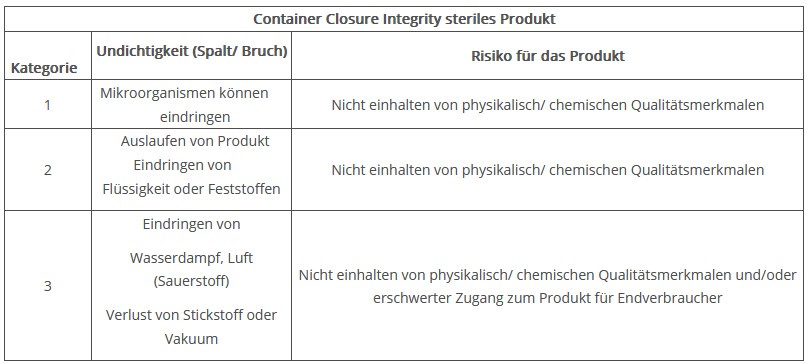
Abbildung 1 Kategorien für Undichtigkeiten (gemäß USP 1207)
Das heißt ein dichtes Verpackungssystem ist nicht per se durch das Bestehen eines mikrobiologischen Challenge Tests oder eines Produkt-Sterilitätstests definiert. Eine dichte Verpackung ist eine, die das maximal zulässige Undichtigkeitslimit (MALL, maximum allowable leakage limit) einhält. Dieses Limit ist immer abhängig von den Anforderungen des Produktes und wird z. B. in mbar*l/s angegeben.
Somit ist klar, dass die Dichtigkeit des Verpackungssystems abhängig ist vom Produkt bzw. von den Anforderungen an dieses Produkt.
Die USP 1207 bildet dazu die in Abbildung 2 dargestellten Kategorien für die Anforderungen an die Qualität der Verpackung:

Abbildung 2 Kategorien für die Anforderungen an die Qualität des Primärverpackungssystems gemäß USP 1207
Die Kategorie 3 stellt eine Subkategorie dar, welche auf Multi-Dose Behältnisse anwendbar ist und in den Kategorien 1 und 2 enthalten sein kann. Bei der Kombination von 2 und 3 bedeutet das, dass sich die Anforderungen an die MALL im Produktzyklus verschieben. Nach der Abfüllung und der Lagerung gilt Kategorie 2, bei der Anwendung gilt aber nur noch die Kategorie 1.
Es gibt einige Untersuchungen dazu, welche Leckage-Raten bei sterilen Produkten anzusetzen sind. Aufgrund der Komplexität soll hier aber nicht weiter darauf eingegangen werden. Für detaillierte Informationen empfiehlt sich ein Studium der USP 1207 bis 1207.3. Generell kann man davon ausgehen, dass für nichtporöse starre Verpackungen wie Vials, Fertigspritzen, Kartuschen, BFS Kunststoffampullen, Ampullen etc. eine Helium leakage rate von < 6 x 10-6 mbar L/s (Kirsch, et all; PDA J Pharm Sci & Technol 51, 5, 1997) ein niedriges Risiko für das Eindringen von Mikroorganismen oder dem Verlust von flüssigem Produkt darstellt. Das entspricht in etwa einer Leckage durch einen Defekt von 0,1 bis 0,3 µm.
Es soll hier nochmals darauf hingewiesen werden, dass die Leckage Rate (MALL) für jedes Produkt/Verpackungssystem spezifisch ist und auf Basis von wissenschaftlichen Daten und des Risikos festgelegt werden muss. Die Kenntnisse der MALL ist wichtig da es verschiedene Arten der Prüfung auf Dichtigkeit gibt und nicht alle Verfahren für jede Undichtigkeit oder MALL gleich gut geeignet sind. Daher sollten, wenn erforderlich, immer mehrere Möglichkeiten der Testung auf Dichtigkeit zur Verfügung stehen.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











