
Konzeptionsphase der Computervalidierung
Auszug aus dem GMP-BERATER, Kapitel 9.E.2.1, Konzeptionsphase
9 Min. Lesezeit | von Dr. Dennis Sandkühler
Erschienen im LOGFILE 27/2022
Der Validierungsplan sollte die Phasen des System-Lebenszyklus abbilden. Sämtliche Aktivitäten der Validierung sollten auf Basis einer fortwährenden Risikobewertung durchgeführt werden.
Änderungen planen
In der Regel gibt es bereits einen definierten Prozess, der durch ein computergestütztes System ergänzt oder durch ein neues System abgelöst werden soll. Im GMP-Umfeld sind diese Änderungen immer durch einen Änderungsantrag (Change Control) zu initiieren. Zum Änderungsantrag gehören u. a.
-
ein Projektantrag (Budget, Zeit, Verantwortlichkeiten),
-
eine initiale Betrachtung der Risiken (durch die Änderung),
-
und ein Projektplan, der u. a. die Erstellung eines Validierungsplans vorsieht.
Validierung planen
Nach Bewilligung der Änderung ist ein Validierungsplan zu erstellen. Einige Unternehmen halten hierzu Vorlagen bereit, die bereits dem Änderungsantrag beigefügt werden.
Die Einführung oder Änderung computergestützter Systeme erfordert ein Zusammenspiel von IT- und QA-Kenntnissen. Änderungen sollten durch die Fachabteilung initiiert und durch Mitwirkung der IT-Abteilung umgesetzt werden. Daher sollten beide Bereiche bereits bei der Erstellung des Validierungsplans zusammenarbeiten. Die IT-Abteilung sollte dabei ebenso wie ein externer IT-Dienstleister qualifiziert sein.
Der Validierungsplan ist im Gegensatz zum Validierungsmasterplan stets bezogen auf ein Validierungsprojekt und beinhaltet daher die konkreten Prüfpunkte und Akzeptanzkriterien des Validierungsprojekts.
Der Aufbau eines Validierungsplans umfasst im Wesentlichen die folgenden Abschnitte:
- Festlegung des Geltungsbereichs und des Validierungsgegenstandes mit einer eindeutigen Bezeichnung des Systems
- Beschreibung des Geschäftsprozesses und Nennung der wesentlichen Funktionen, die das computergestützte System unterstützt
- Systemübersicht mit Schnittstellen und Abhängigkeiten zu anderen Systemen
- Validierungsplanung und damit Festlegung der anstehenden Validierungsaktivitäten und Regelung der Verantwortlichkeiten
- Festlegung der Prüfpunkte und Akzeptanzkriterien
- Festlegung der Akzeptanzkriterien für eine erfolgreiche Validierung
- Definition des Abschlusses der Validierung und Inbetriebnahme
Projektrollen definieren und Verantwortlichkeiten festlegen
Für den Erfolg eines jeden Projekts ist es wichtig, die Projektorganisation und Verantwortungen von Anfang an festzulegen und zu kommunizieren.
Die Verantwortlichkeiten können dabei in einer Verantwortlichkeitsmatrix dargestellt werden. Die RACI-Matrix stellt eine mögliche Technik zur Visualisierung von Verantwortlichen dar:
- R – Responsible (verantwortlich)
- Person, die unmittelbar mit der Aktivität betraut ist. Es sollte immer nur eine verantwortliche Person pro Aufgabe geben.
- A – Accountable (rechenschaftspflichtig)
- Person, die verantwortlich ist, dass die Aktivität erledigt wird; z. B. der Projektmanager.
- C – Consulted (konsultiert)
- Personen, die unterstützend beteiligt sind oder über weitere Informationen zur Aktivität verfügen.
- I – Informed (informiert)
- Personen, die über den aktuellen Stand oder den Abschluss einer Aktivität informiert werden sollten.
Die RACI-Matrix kann die Prüfpunkte und Akzeptanzkriterien sehr gut ergänzen, entweder als eigenständige Matrix mit Auflistung der Aktivitätsnummer oder zusammengefasst in einer Matrix.
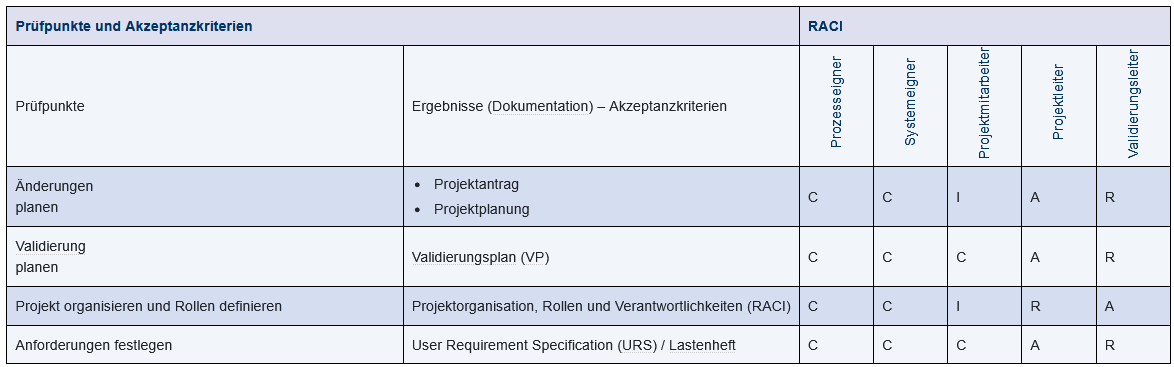
Abbildung 9.E-11 Kombinierte Validierungs- und Verantwortungsmatrix
Anforderungen festlegen (Lastenheft)
In einem Lastenheft (oder auch User Requirement Specification, URS genannt) müssen allen regulatorischen und Prozess-Anforderungen zusammengestellt werden. Hier ist es hilfreich, zunächst alle regulatorischen Anforderungen zu erfassen und anschließend die Anforderungen an den Prozess in einem eigenen Abschnitt zusammen zu stellen.
Die Prozessanforderungen sollten klar und unmissverständlich formuliert sein und sollten sich nur auf den jeweiligen Geschäftsprozess beziehen.
Um im Rahmen der Validierung erreichbar und überprüfbar zu sein, sollten die Anforderungen SMART formuliert sein, d. h. spezifisch, messbar, akzeptiert, realistisch und terminiert.

Das Lastenheft ist nicht statisch und kann sich durch ergänzende Eingaben und insbesondere durch die Risikobewertung um weitere Anforderungen ergänzen.
Technische und funktionale Umsetzung prüfen (Pflichtenheft)
Nach der Festlegung des Lastenhefts erfolgt die Prüfung der technischen und funktionalen Umsetzung in einem Pflichtenheft. Das Pflichtenheft wird auch als Functional Specification (FS) bezeichnet. Die Prüfung erfolgt im Allgemeinen durch einen Anbietervergleich (Anbieter = Lieferant). Je SMARTer die Formulierung der Anforderungen, desto eindeutiger und schneller erfolgt die Bewertung, ob die Anforderungen erfüllt werden.
Bei individueller bzw. Eigenentwicklung von Hard- und Software sind hierbei auch Entwicklungsrichtlinien festzulegen und zu überwachen, auf die hier nicht näher eingegangen wird.
Lasten und Pflichten (oder URS und FS) sollten in einer Traceability Matrix gegenübergestellt werden. So lassen sich die Anforderungen gut überblicken und nicht erfüllte Anforderungen schnell identifizieren. Zudem können auch weitere Werte und Entscheidungen oder Anmerkungen die Traceability Matrix ergänzen. Bei Soft- und Hardware (z. B. Softwarekategorie 3 und 4) verfügt der Lieferant in der Regel über einen Katalog an spezifizierten Funktionen (Functional Specification), die seine Software oder Hardware erfüllt. In der Traceability Matrix kann auf diese Functional Specification referenziert werden. Abbildung 9.E-12 zeigt beispielhaft den Aufbau einer Traceability Matrix.
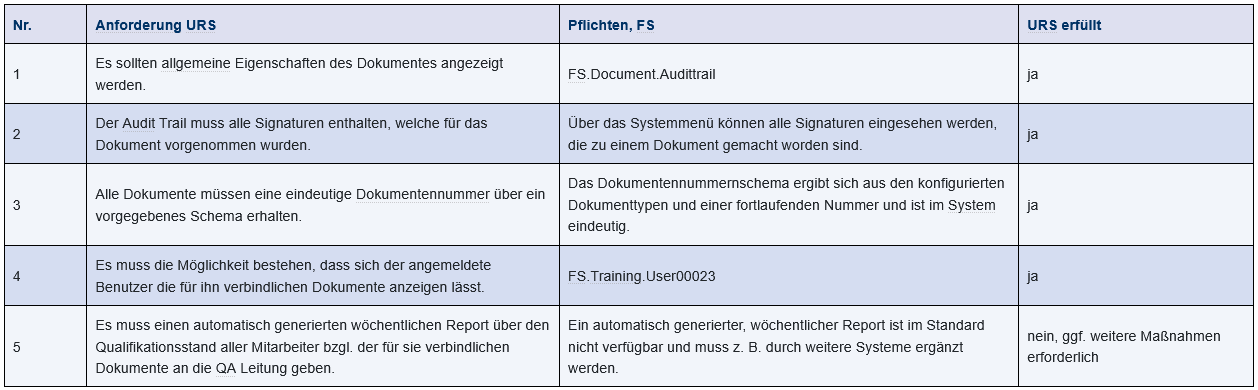
Abbildung 9.E-12 Beispiel: Lasten- und Pflichtenheft zur Einführung eines computergestützten Dokumentenmanagementsystems (Traceability Matrix)
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











