

Kritikalitätseinstufungen von Abweichungen
Ein Vorgehen nach der PIC/S Guidance on Classification of GMP Deficiencies
5 Min. Lesezeit | von Dr. Felix Tobias Kern und Liwa Schneider
Erschienen im LOGFILE 40/2020
Die korrekte und konsequente Einstufung von Abweichungen in die Kategorien Minor, Major und Critical stellen pharmazeutische Hersteller immer wieder vor Herausforderungen. Eine systematische Vorgabe ist Voraussetzung für das Eliminieren von subjektiven Komponenten der einstufenden QA Funktion und der QPs. Auch verhindert diese Vorgabe Inhomogenitäten zwischen den einzelnen Entscheidungsträgern der QA und zwischen den einzelnen QPs.
Die PIC/S Guidance on Classification of GMP Deficiencies kann hierzu als Orientierungshilfe dienen.
Diese PIC/S Guidance dient eigentlich zur Unterstützung von GMP-Inspektoren, um eine risikobasierte und harmonisierte Klassifizierung von GMP-Mängeln zu ermöglichen. Da es sich bei Abweichungen in der Herstellung weitestgehend um GMP-Mängel handelt, kann diese Guidance auch als Grundlage für ein Klassifizierungssystem von Abweichungen herangezogen werden.
Definitionen Minor, Major und Critical nach der PIC/S Guidance
Die Guidance unterscheidet drei Klassen von GMP-Mängeln: „Critical Deficiency“, „Major Deficiency“ und „Other Deficiency”.
Critical
Die kritische Abweichung resultiert in einem Produkt, welches den Patienten gefährdet oder ein signifikantes Risiko hierfür darstellt. Der Zeitpunkt der Entdeckung, beispielweise noch während der Produktion, vor oder schon nach der Freigabe, und die Entdeckungswahrscheinlichkeit hierfür werden nicht betrachtet. Wird also eine Charge mit einem Qualitätsmangel produziert, der den Patienten gefährdet, der Hersteller dies aber durch repräsentative oder automatische Kontrollsysteme bemerkt, ist der Guidance folgend dieser Mangel und damit die Abweichung als „Critical“ einzustufen.
Major
Ein „Major Deficiency“- ist eine Abweichung, die kein „Critical Deficiency“ ist. Das bedeutet, dass der Patient durch diesen Mangel/durch die Abweichung nicht gefährdet ist und es besteht auch kein Risiko hierfür. Beispiele für „Major“-Abweichungen sind:
- Verstöße gegen Zulassungsdokumente und Spezifikationen, gegen die Herstellungserlaubnis, gegen die Genehmigung zur klinischen Prüfung oder gegen Arzneibücher
- ineffektive Umsetzung der geforderten GMP-Kontrollen, z. B. IPC-Kontrollen oder weitere Kontrollen während der Produktion, beispielsweise zu Aufdrucken von Chargendaten auf Faltschachteln, sowie der Batch Release Test,
- unzuverlässiges Freigabeprozedere: Dies betrifft sowohl Abweichungen bezüglich des Systems an sich, z. B. die Freigabe von Chargen ohne abgeschlossene Untersuchung einer aufgetretenen Abweichung, als auch der für die Freigabe verantwortlichen Personen.
Other (Minor)
Die Defintion von „Other Deficiency“ entspricht nicht der Definition der ersten beiden Kategorien, aber trotzdem wird von den Regeln der Good Manufacturing Practice (GMP) abgewichen. Man kann diese Klasse auch als „Minor Deficiency“ bezeichnen.
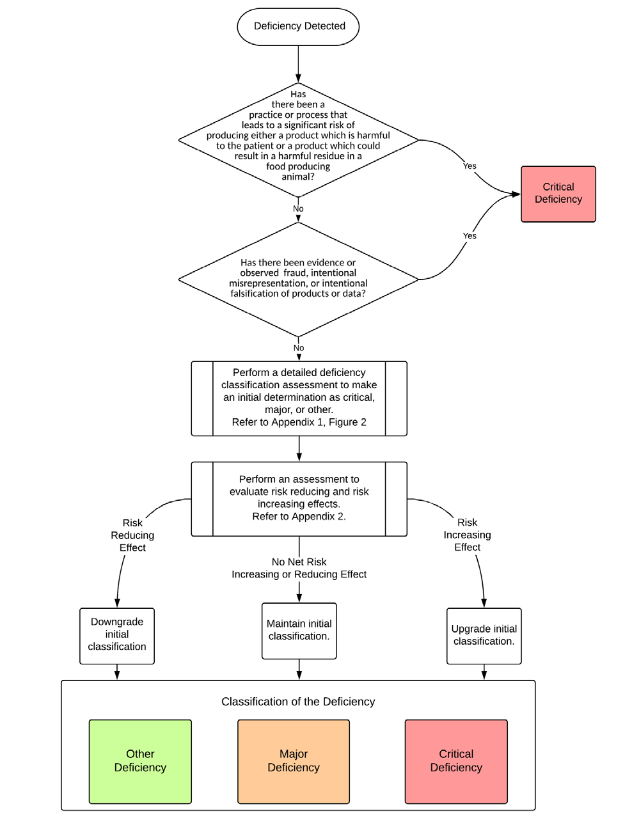
Risikoerhöhende und risikominimierende Faktoren, die die initiale Klassifizierung verändern können.
Die Guidance führt zusätzlich Faktoren an, die die initiale Risikoklasse final erhöhen oder erniedrigen können.
Zu den risikoerhöhenden Faktoren zählt beispielsweise das wiederholte Auftreten einer Abweichung, auch Recurring genannt. Als Recurring-Abweichungen können Abweichungen verstanden werden, die erneut an derselben Linie oder an demselben Equipment aufgetreten sind und bei denen CAPA-Maßnahmen nicht verhindern konnten, dass die Abweichung erneut aufgetreten ist. Sollten sich im Produktionsbetrieb eine zweite vergleichbare Linie befinden oder ein vergleichbares Equipment, ist dies mit in die Betrachtung einzubeziehen.
Risikoreduzierende Faktoren richten sich auf das Thema Risikomanagement. Wurden CAPA-Maßnahmen auf Basis der Abweichung initiiert oder bereits etabliert, die das Produkt-, das Patientenrisiko und/oder weitere Risiken reduzieren?
Zusammenfassung
Die PIC/S Guidance on Classification of GMP Deficiencies bietet für einen Herstellbetrieb eine geeignete Rationale, Klassifizierungsregeln für Abweichungen aufzustellen. Die Guidance enthält zahlreiche praxisrelevante Beispiele und Entscheidungsbäume. Weiterhin findet sich eine Liste an risikoerhöhenden und risikominimierenden Faktoren, mit denen die Risikobewertung nochmals justiert werden kann.
Überblick über den Klassifizierungsprozess von GMP-Mängeln (Quelle: PIC/S Guidance on Classification of GMP Deficiencies, Appendix 1, Figure 1)
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











