

Lastenheft für eine Anlagensoftware GMP-konform erstellen
Auszug aus dem GMP:KnowHow Anlagenqualifizierung
8 Min. Lesezeit | von Petra Berlemann
Erschienen im LOGFILE Leitartikel 23/2022
Stellen Sie sich vor, Sie müssen in eine bestehende pharmazeutische Abfüllanlage in ca. sechs Monaten eine neue Software zur vollautomatisierten Abfüllung von flüssigen Substanzen implementieren. Als erstes brauchen Sie eine kurze Projektbeschreibung und entwickeln daraus Benutzeranforderungen / ein Lastenheft.
Das Lern- und Wissensportal GMP:KnowHow Anlagenqualifizierung hilft Ihnen dabei mit einem beispielhaften Aufbau, wie ein Lastenheft für eine Software aussehen könnte.
In einem Auszug aus dem Portal erfahren Sie mehr zu möglichen Leitfragen und Benutzeranforderungen.
Benutzeranforderung (BA), User Requirement Specification (URS) und Lastenheft: Was ist das? Wie erstellt man solche Dokumente? Ist das alles dasselbe? Oder doch etwas ganz anderes?
Kurz: Alle Ausdrücke beschreiben die Anforderungen an ein Produkt, zum Beispiel Software aus Benutzersicht. Üblicherweise erstellt man sie bei einer Neuanschaffung oder Änderung an einem bestehenden System mit folgenden Inhalten:
- Was muss diese Software können?
- Was erwarte ich von ihr?
- Welche Schnittstellen sind zu beachten?
- Wer schreibt eine Benutzeranforderung?
Dieses sind die Leitfragen bei der Erstellung einer Benutzeranforderung oder auch eines Lastenhefts.
Eine Benutzeranforderung wird von dem Personenkreis verfasst, der die Software oder das Computersystem benutzen wird. Es werden die Anforderungen beschrieben und nicht die fertige Lösung. Die verschiedenen Anforderungen dürfen sich nicht widersprechen. Die Benutzeranforderungen können in tabellarischer Form oder in Fließtext beschrieben werden. Der Vorteil der tabellarischen Form ist die Übersichtlichkeit und die Zuordnung einer Nummer zu einer Benutzeranforderung. Die Traceability (Rückverfolgbarkeit) von Benutzeranforderungen ist dadurch gegeben. Nachteilig für die tabellarische Darstellung ist die kurze Beschreibung der Benutzeranforderungen.
Beispiel: Ich möchte ein Handschreibgerät mit dokumentenechter Tinte. Die Tinte muss blau sein, und das Handschreibgerät darf nicht mehr als 100 g wiegen, eine Gesamtlänge von max. 20 cm aufweisen und im Durchmesser ca. 15 mm stark sein.
Nicht: Ich möchte einen Kugelschreiber der Marke XYZ1, der blau schreibt. Das wäre bereits die Lösung.
Nachfolgend ein beispielhafter Aufbau mit Beispielen, wie eine Benutzeranforderung für eine Software/ein Computersystem aussehen könnte. Die verschiedenen Kapitel sind beliebig ausbaubar, je nach eigener Anforderung.
Zuerst startet man mit einer kurzen Projektbeschreibung. Die Benutzeranforderung wird im Normalfall an einen oder mehrere potentielle(n) Lieferanten gesendet. So wissen sie, worum es geht, und können eine passende Lösung vorschlagen.
Dieser Lösungsvorschlag vom Lieferanten wird Functional Specification oder Funktionsspezifikation genannt.
Mögliche Leitfragen zu einer Projektbeschreibung könnten sein:
- Was möchte ich meinem potentiellen Lieferanten mitteilen?
- Um was geht es eigentlich im Projekt?
- Wo ist der Ort, an dem die Software eingesetzt wird?
Beispiel Projektbeschreibung
In eine bestehende pharmazeutische Abfüllanlage soll in ca. 6 Monaten eine neue Software zur vollautomatisierten Abfüllung von flüssigen Substanzen implementiert werden. Die Abfüllanlage ist im Reinraum der Klasse 123XX untergebracht. Diese Abfüllanlage wird von ca. 5 Mitarbeitern bedient. Das Bedienpanel befindet sich im Reinraum, und die gesamte Softwareanbindung ist in der Prozess-Schaltzentrale implementiert. Die Lieblingsfarbe der Mitarbeitenden ist Neonpink. Vom Bedienpanel kann ausgedruckt werden. Die Anzeigen des Bedienpanels werden ausschließlich zu Informationszwecken genutzt. Die Datenverarbeitung und Datenübertragung sowie Datensicherung werden elektronisch (digital) vorgenommen.
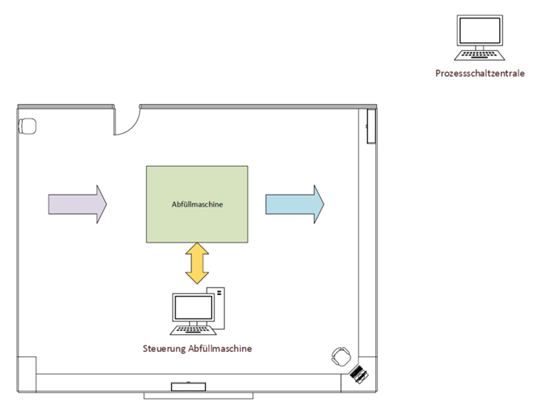
Abbildung 1 – Skizze Abfüllmaschine im Reinraum
Wer jetzt aufmerksam gelesen hat, hat bemerkt, dass sich in der Projektbeschreibung bereits Anforderungen verstecken. Anforderungen müssen für ein erfolgreiches Projekt unbedingt eingehalten werden. Die Abgrenzung zwischen einer Beschreibung und einer Anforderung ist sehr differenziert und oft schwierig.
Die versteckten Anforderungen in der Projektbeschreibung sind kategorisierbar. Es gibt „Muss“-, „Kann“- und „Nice-to-have“-Anforderungskategorien. Ebenso wäre es möglich, zusätzlich in GMP-Anforderungen und Nicht-GMP-Anforderungen zu kategorisieren.
Mögliche Leitfragen zur Kategorisierung wären beispielsweise:
Werden alle Anforderungen unbedingt zum erfolgreichen Abschluss des Projektes benötigt?
Welche müssen in jedem Fall erfüllt werden?
Welche können erfüllt werden, und welche Anforderungen wären einfach wünschenswert – eben ein „Nice-to-have“?
Für die in der obigen Projektbeschreibung versteckten Anforderungen könnte wie in Abbildung 2 dokumentiert werden.
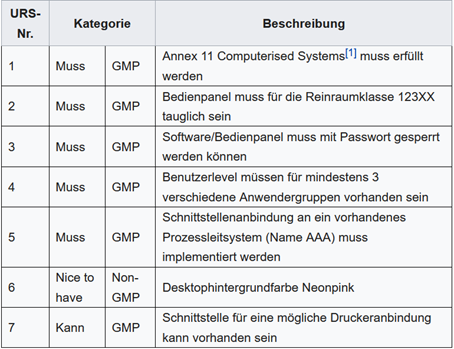
Abbildung 2 – Benutzeranforderungen unsortiert
Es ist empfehlenswert, eine grobe Struktur des Dokumentes vorzubereiten, damit nichts vergessen wird. In Abbildung 2 sind alle Anforderungen unstrukturiert aufgelistet: Richtlinien, Software, Schnittstellen. So behält man leicht den Überblick.
In den Abbildungen 3 bis 6, welche Sie im Wissensportal GMP:KnowHow Anlagenqualifizierung finden können, werden mögliche Anforderungskategorien zusammengefasst, diese sind nicht abschließend. Auch die Nummerierung kann nach persönlicher Präferenz durchgeführt werden. Hier gibt es kein Falsch oder Richtig. Die URS-Nummern sind verschiedenen Kategorien zugeordnet.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











