

Leistungsfähigkeit des CAPA-Systems
7 Min. Lesezeit | von Dr. Christian Gausepohl
Erschienen im LOGFILE Leitartikel 43/2023
Der aktuelle Entwurf der FDA zu Qualitätskennzahlen bzw. Reifegrad der Qualitätssysteme berücksichtigt auch die Leistungsfähigkeit des CAPA-Systems als einen Indikator für das Pharmazeutische Qualitätssystem.
Die Überwachung der Leistungsfähigkeit (Governance) des CAPA-Systems im Unternehmen ist eine Aufgabe des Managements, das für das Pharmazeutische Qualitätssystem verantwortlich ist. Diese Verantwortlichkeit und die Vorgehensweise müssen in den entsprechenden Verfahrensanweisungen beschrieben sein.
Der Blick auf das CAPA-Management und seine Leistungsfähigkeit liefert zudem einen guten Einblick auf die Qualitätskultur (Quality Culture) des Unternehmens. Im Rahmen einer Inspektion wird besonders auf wiederholt auftretende Fehler, nicht eingehaltene CAPA-Zieltermine und Effektivitätsprüfungen geachtet. Hiervon ausgehend kann direkt in mögliche Problembereiche abgetaucht werden.
Das CAPA-Management ist Teil der kontinuierlichen Verbesserung von Produkten und Prozessen. Die Effektivität und Effizienz des CAPA-Managements werden durch mehrere Faktoren beeinflusst:
A Governance
Um diese Managementverantwortlichkeit sinnvoll wahrnehmen zu können, sind verschiedene Teilaspekte von Bedeutung:
- Organisation und Führungsverhalten
Die organisatorischen Zusammenhänge und Verantwortlichkeiten für die Überwachung des CAPA-Management müssen klar beschrieben sein inkl. der Prozesse für Reporting, Genehmigung und Eskalation. Daneben spielt das Führungsverhalten eine wichtige Rolle, in dem die Bedeutung vom Umgang mit Fehlern, Maßnahmen und kontinuierlicher Verbesserung unterstrichen und aktiv verankert werden kann.
- CAPA-Monitoring
Die regelmäßige Überprüfung der Effektivität des CAPA-Management berücksichtigt CAPA-spezifische KPIs mit Trendanalysen, Bewertung der Ressourcen und Kompetenzen im Unternehmen.
B Qualitätskultur
Qualität ist nicht eine Frage einzelner Personen, z. B. Qualitätsmanager, oder der Qualitätsabteilungen wie Qualitätssicherung, Qualitätskontrolle oder der Sachkundigen Person, sondern jedes einzelnen Mitarbeiters – jeder ist für Qualität mitverantwortlich. Qualitätsdenken ist Einstellungssache jedes Einzelnen, beginnt an der Spitze der Organisation und muss auf allen Ebenen übernommen werden. Das Qualitätsbewusstsein und den Willen zur kontinuierlichen Verbesserung zu entwickeln und zu pflegen, ist eine gemeinsame Führungsverantwortlichkeit. Dabei gilt es nicht, Fehler oder gar den Verursacher in den Mittelpunkt zu stellen, sondern deren Vermeidung.
Die FDA hat zu einer regelmäßigen Bewertung der Qualitätskultur durch das Management aufgerufen. Hierzu haben verschiedene Organisationen Methoden entwickelt, z. B. das Quality Culture Assessment Tool and Training der PDA oder das Advancing Pharmaceutical Quality Program der ISPE. Diese Methoden bewerten den aktuellen Reifegrad des Qualitätssystems und erlauben dessen kontrollierte Weiterentwicklung.
Im Sinne der kontinuierlichen Verbesserung können verschiedene Phasen mit Blick auf Verbesserungsmaßnahmen durchlaufen werden (siehe auch Abbildung 1):
- Reaktive Phase
In dieser Phase überwiegen die Korrekturmaßnahmen basierend auf bestehenden Fehlern. Die Ursachen für die Fehler werden nach und nach eliminiert und dadurch die Gesamtanzahl der Fehler reduziert.
- Proaktive Phase
In die Zukunft gerichtet sind die Vorbeugemaßnahmen, die in dieser Phase im Vordergrund stehen und für eine fortlaufend weitere Fehlerreduktion sorgen. Diese Phase spiegelt einen verbesserten Reifegrad der Qualitätskultur wider.
- Six Sigma Phase
Hier geht es im Schwerpunkt nicht mehr um Fehler und deren Vermeidung, sondern um die kontrollierte, fortlaufende Reduktion von Prozessvariabilitäten. Der Reifegrad der Qualitätskultur ist weiter verbessert.
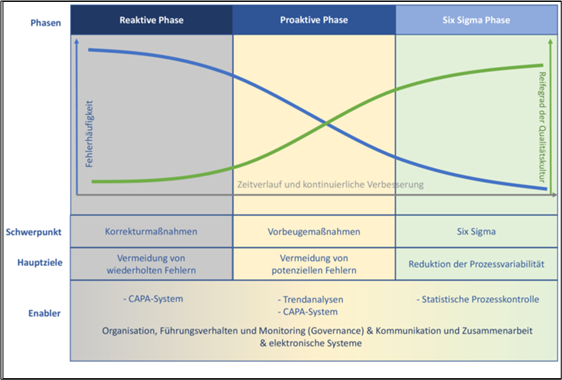
Abbildung 1 CAPA-Management im Rahmen der kontinuierlichen Verbesserung
C Kommunikation und Zusammenarbeit
Die offene und transparente Kommunikation zu Fehlern, Fehlerursachen und Maßnahmen sowie deren Bedeutung unterstützt das Qualitätsbewusstsein auf breiter Front. Eine offene und gute Zusammenarbeit ist grundsätzlich eine wichtige Säule der Qualitätskultur und trägt zu einem leistungsfähigen CAPA-Management bei.
D Elektronische Systeme
Elektronische Systeme im CAPA-Management können wesentlich zur Verbesserung von Effektivität und Effizienz beitragen. Die Daten liegen transparent vor und können im Unternehmen den Verantwortlichen einfach und standardisiert zur Verfügung gestellt werden. Automatisierte Reports können, z. B. wöchentlich, direkt an das Management oder die Fachbereichsverantwortlichen geschickt werden ohne einen gesonderten Filter durch die Qualitätssicherung. Auch hier halten automatisierte, selbstlernende Systeme und künstliche Intelligenz (AI) Einzug, die Daten in der Analyse sinnvoll verknüpfen.
| Die Leistungsfähigkeit des CAPA-Management ist Gegenstand jeder Inspektion. Die CAPA-KPIs erlauben Rückschlüsse auf die Qualitätskultur im Unternehmen. |
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











