

Medizinischer Cannabis und GMP
8 Min. Lesezeit | von Marta Rodríguez
Erschienen im LOGFILE 40/2022
Kapseln, Tropfen oder Joint – von der Vielfalt pflanzlicher Arzneimittel! – Die neue Folge von unserem Webcast GMP & TEA beleuchtet die Besonderheiten pflanzlicher Arzneimittel. Thomas Peither erläutert unter anderem, ab wann die GACP- und die GMP-Anforderungen greifen und warum Cannabis anders ist.
Im heutigen Leitartikel dreht sich alles um medizinischen Cannabis und GMP. Marta Rodríguez geht ebenfalls insbesondere auf die Frage ein, ab wann GACP und ab wann GMP anzuwenden ist.
Der Anbau, die Herstellung, die Vermarktung, die Einfuhr und die Ausfuhr von Zubereitungen und Substanzen auf der Grundlage der Cannabispflanze zu medizinischen und Forschungszwecken unterliegen der Kontrolle durch die zuständigen Behörden.
Die für die Produktion, die Kontrolle, den Vertrieb und die Abgabe zugelassenen Stellen sowie die Art der Produkte, die Vertriebskanäle und die beabsichtigten Verwendungszwecke hängen in hohem Maße von den nationalen Vorschriften ab, die in den meisten Ländern derzeit noch entwickelt und implementiert werden.
Der Anbau, die Verarbeitung nach der Ernte (Zerschneiden, Trocknen, Aushärten, Verpacken) und die Extraktion/Reinigung des Wirkstoffs (THC, CBD oder andere Cannabinoide) müssen in der EU unter Einhaltung der GACP- (Good Agricultural and Cultivation Practices) und den GMP-Leitlinien (Good Manufacturing Practices) für die weiteren Verarbeitungs- und Handhabungsstufen erfolgen.
Der Punkt, ab dem die GMP-Standards angewendet werden sollten, ist jedoch ein häufiger Diskussionspunkt. Hinweise finden sich beispielsweise im EU-GMP-Leitfaden Teil II oder im EU-GMP-Leitfaden Anhang 7. Ein risikobasierter Ansatz ist der Schlüssel zur Ermittlung des ersten kritischen Schritts des Produktionsprozesses, ab dem die GMP-Standards angewendet werden sollten.
Die Art des Endprodukts, das bei der Herstellung entsteht, bestimmt das Risiko der vorangegangenen Schritte. Je mehr Prozessschritte es gibt, desto später sollte mit der Anwendung von GMP-Standards begonnen werden. Handelt es sich bei dem Endprodukt um die getrocknete Blüte, sollten die GMP-Standards bereits bei den ersten Tätigkeiten nach der Ernte (Zerschneiden, Trocknen und Aushärten) angewendet werden. Handelt es sich bei dem Endprodukt jedoch um den gereinigten THC- oder CBD-Extrakt, dann könnte der erste kritische Schritt das Mahlen der getrockneten Blüten sein, bevor der Extraktionsprozess gestartet wird. Es ist eine Einzelfallanalyse erforderlich, bei der der gesamte Prozess und seine Risiken berücksichtigt werden, damit eine solide Bewertung der spezifischen Anforderungen vorgenommen werden kann.
Die folgenden Abbildungen zeigen beispielhaft für zwei verschiedene Konstellationen, ab wann GACP- und GMP-Anforderungen anzuwenden sind.
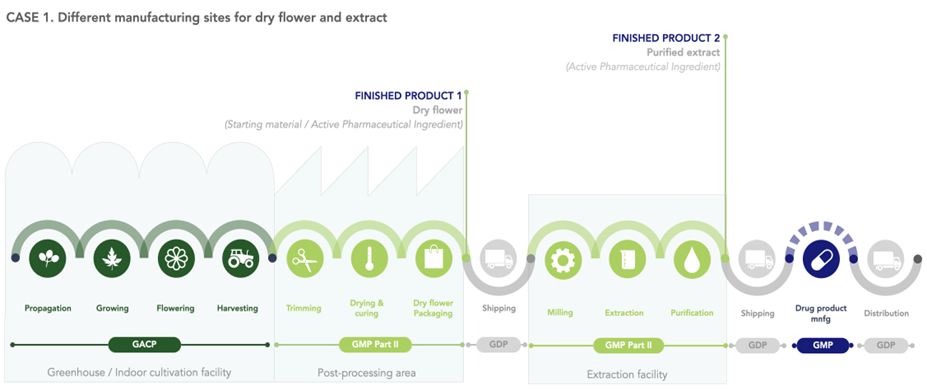
Abbildung 1 Ab wann sind die GMP-Standards anzuwenden? Fall 1 - verschiedene Herstellungsstätten für die getrockneten Blüten und den Extrakt
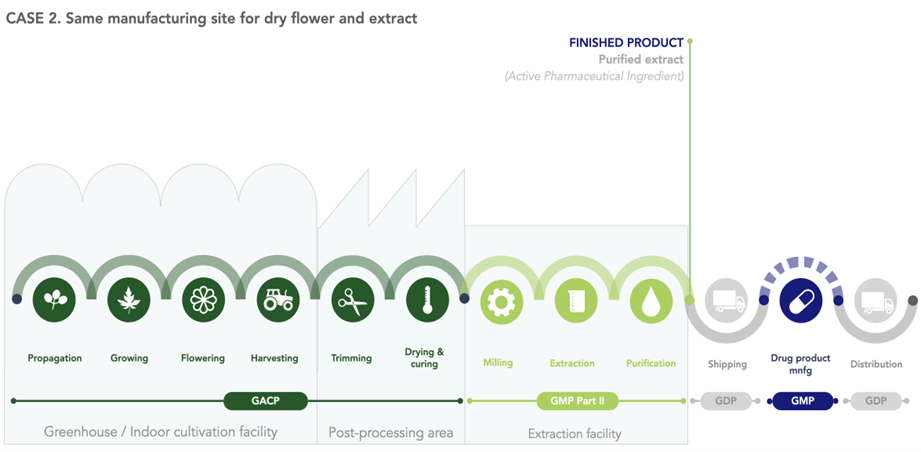
Abbildung 2 Ab wann sind die GMP-Standards anzuwenden? Fall 2 – dieselbe Herstellungsstätte für die getrockneten Blüten und den Extrakt
GMP und medizinischer Cannabis
Nach der Festlegung des Geltungsbereichs für die GMP-Anforderungen müssen die GMP-Anforderungen identifiziert und definiert werden, wie sie im jeweiligen Projekt erfüllt werden sollten. Die EU-GMP-Regularien werden in jedem EU-Land von der zuständigen Gesundheitsbehörde national umgesetzt, ggf. mit spezifischen Anforderungen und unterschiedlichen Anforderungsniveaus. Einige Leitlinien und Standards werden auch von internationalen Harmonisierungsgremien wie der ICH herausgegeben, und zwischen
verschiedenen Gesundheitsbehörden sind Vereinbarungen über die gegenseitige Anerkennung getroffen worden.
In Europa veröffentlicht die Europäische Kommission den EU-GMP-Leitfaden sowie ergänzende Verordnungen und Leitlinien, die dann von den nationalen Gesundheitsbehörden in den Rechtsrahmen der einzelnen Länder übertragen werden.
Der EU-GMP-Leitfaden gliedert sich in vier Teile, 21 Anhänge und weitere GMP-bezogene Dokumente. Teil I enthält die Vorschriften für Fertigarzneimittel, und Teil II gilt für pharmazeutische Wirkstoffe (API). Die Anhänge enthalten spezifische Vorschriften, die für eine bestimmte Tätigkeit oder eine bestimmte Art von Produkt gelten, wie z. B. Anhang 7, der spezielle Hinweise für Arzneimittel auf der Grundlage von Pflanzenextrakten enthält, was für Arzneimittel basierend auf Cannabis sativa relevant ist.
Darüber hinaus sind bei pflanzlichen Arzneimitteln die Auswahl des Saatguts sowie die Anbau- und Erntebedingungen Aspekte, die einen großen Einfluss auf die Qualität der Pflanze und damit auf die Qualität des Wirkstoffs haben. Der Leitfaden des Ausschusses für pflanzliche Arzneimittel (HMPC) der EMA "Guideline on Good Agricultural and Collection Practice (GACP) for Starting Materials of Herbal Origin" enthält Empfehlungen für die Einführung eines Qualitätssicherungssystems.
Abbildung 3 zeigt die sechs wichtigsten Faktoren, die zur GMP-Konformität beitragen.
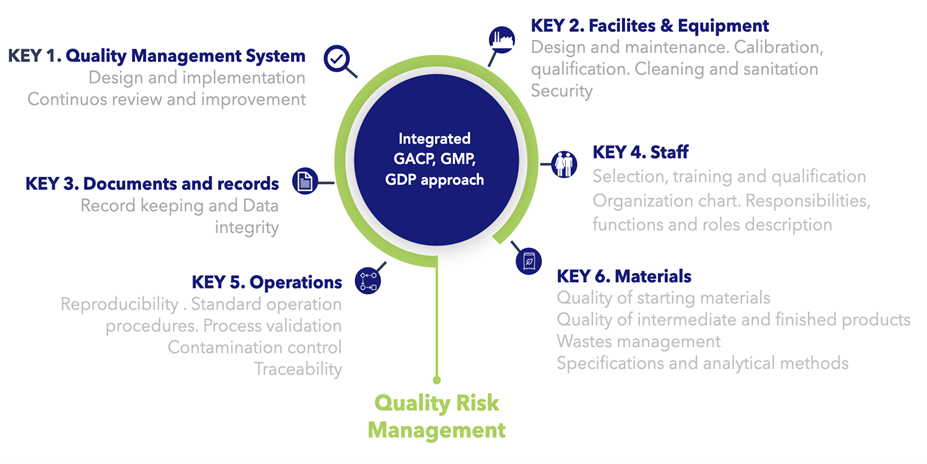
Abbildung 3 6 Faktoren zur GMP-Konformität
Was Sie im Einzelnen zu den 6 Faktoren berücksichtigen müssen, lesen Sie im vollständigen Kapitel 0.B.2 des internationalen GMP-Wissensportals, dem GMP Compliance Adviser. Dieser Text ist ein übersetzter Auszug aus dem Kapitel 0.B.2 Medical Cannabis and GMP: 6 Keys to Focus on.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











