

Mindeststandards für die Prüfung von Softwareentwicklung
Ein Auszug aus dem GMP-Fachwissen Computergestützte Systeme im GMP-Umfeld, Kapitel 3.D
9 Min. Lesezeit | von
Markus Roemer und Dr. Siegfried Schmitt
Erschienen im LOGFILE 09/2022
Es gibt verschiedene Methoden, Standards und Ansätze für die Entwicklung von Software. Diese sind – mit Abstrichen und Anpassungen – analog auch für Hardware-Produkte anwendbar.
Der ISPE GAMP®5-Leitfaden verweist zum Beispiel auf verschiedene Standards wie
- CMMI (Capability Maturity Model Integration),
- RUP (Rational Unified Process),
- RAD (Rapid Application Development).
Daneben gibt es weitere agile Methoden oder Normen wie ISO 12207 und SPICE (Software Process Improvement and Capability Determination1).
Im V-Modell wird die Entwicklung in einem einzigen Block dargestellt, in dem sich jedoch ein komplettes Eigenleben abspielt. Die oben genannten Standards und Normen umfassen mehrere hundert Seiten und sind inhaltlich sehr breit aufgestellt. Als Qualitäts- oder Validierungsbeauftragter sollte man eine Wissenstiefe in diesen Themen anstreben, die es einem erlaubt, solche Methoden bewerten zu können, ohne alle Details kennen zu müssen.
Der sogenannte Softwareentwicklungs-Lebenszyklus, aus dem übrigens auch die Validierung ihr V-Modell übernommen hat, beschreibt den Weg einer eingereichten Spezifikation über die Entwicklung zum Entwicklertest in das fertige Software Produkt (Release). In aller Regel werden komplexere Entwicklungstätigkeiten durch mehrere Entwickler umgesetzt. Daher ist es nötig, diesen Arbeitsfluss und die Methoden zu beschreiben.
Der Lieferant eines Systems kann sowohl firmenintern, durch eine externe Fachfirma oder durch einen Generalunternehmer repräsentiert sein. Durch Annahme der Benutzeranforderungen (Spezifikationen) geht der Lieferant (durch entsprechende vertragliche Bindungen) eine Bringschuld ein.
Sonderfall Open-Source-Software
Bei sogenannter „Open-Source-Software“ ist ein Verantwortlicher zu definieren, der die Lieferantenverantwortung hierfür übernimmt. Bei einer koordinierten Administration und Kontrolle kann diese Art der Software auch im regulierten Bereich verwendet werden.
Bei solchen Softwareprodukten arbeiten viele, generell unbekannte Entwickler mit. Ein Audit des Lieferanten im klassischen Sinne ist hier nicht möglich. Dieses Manko kann beispielsweise durch intensivere Prüfung ausgeglichen werden; dies insbesondere auch bevor Patches oder Upgrades installiert werden. Der Nachweis des korrekten Funktionierens der Software im GMP-Umfeld ist essentiell, wie auch die permanente Kontrolle über den Zugriff auf die Software. Wann und wo Open-Source-Software in einer Firma eingesetzt werden darf, sollte in internen Anweisungen und Risikoanalysen klar geregelt und analysiert werden. Die Anweisungen zur Computersystemvalidierung sollten diesen Aspekt beinhalten.
Annex 11 fordert, dass der Lieferant ein Qualitätsmanagementsystem (QMS) vorweisen muss, unabhängig davon, ob der Lieferant eine interne Abteilung oder ein externer Dienstleister, ein Klein- oder Großunternehmen ist. In diesem QMS muss zwingend der Softwareentwicklungs-Lebenszyklus definiert und integriert sein. Der pharmazeutische Betreiber verschafft sich während des Lieferantenaudits einen Einblick in dieses QMS und die Softwareentwicklungsmethodik. Das ausgewählte Auditteam kann dabei aus mehreren Personen bestehen, die verschiedene Wissensgebiete abdecken können. Ob ein Audit vor Ort oder aus der Ferne durchgeführt wird, hängt von verschiedenen Faktoren ab. Sinnvollerweise, sollte der Entscheid in einer Risikoanalyse oder einem anderen geführten Dokument zusammen mit der Rationale dokumentiert werden.
Die Entwicklungsmethodik ist sehr technisch ausgerichtet und sollte (neben allgemeinen Qualitätsmanagementaspekten) mindestens folgende Standards für eine Prüfung beinhalten (siehe auch Abbildung 1):
- Standards für die Programmierung: Programmierrichtlinien und einheitliche Konventionen (z. B. Variablennamen, Kommentardichte und Vermerke von Änderungen) müssen vorgegeben sein. Die verwendeten Entwicklungswerkzeuge und die Entwicklungsumgebung müssen verifiziert und festgelegt sein. Die Verwendung von Codes/Applikationen von Drittanbietern (Third Party Code) muss geregelt sein.
- Codierung: Für die Umsetzung der Programmierung werden einzelne Entwicklungsaufträge auf Grundlage der Anforderungen und gewünschten Funktionen in Codesegmente oder Units für die Entwickler generiert.
- Source-Code-Management: Das Source-Code-Management ist eine definierte Ablage des Quellcodes durch die Entwickler. Hier können sogenannte Repository Management Systeme zum Einsatz kommen. Eine Software-Applikation besteht in der Regel aus mehreren hundert Dateien, die während der Entwicklung erstellt oder modifiziert werden – und dementsprechend verwaltet werden müssen.
- Datensicherung: Der Source-Code muss entsprechend gesichert werden, sowohl während der Entwicklung als auch während des operativen Betriebs.
- Testmanagement: Die Entwickler und/oder die interne Qualitätsabteilung führen Tests der Software in verschiedenen Phasen durch. Die Teststrategie, Umgebung, Planung, Durchführung, Dokumentation und Berichterstattung müssen geregelt sein.
- Fehlerbehandlung: Während der Projekt- und Betriebsphase muss ein Prozess für die Fehlerbehandlung definiert sein.
- Release-Management: Für das Release-Management des Produkts müssen Verfahren und Dokumente vorhanden sein, die regeln, wie das Produkt weiterentwickelt wird und wie lange die Wartung sichergestellt ist.
- Schulung: Das Schulungsverfahren regelt, wie die Entwickler in den o. g. Themen geschult werden.
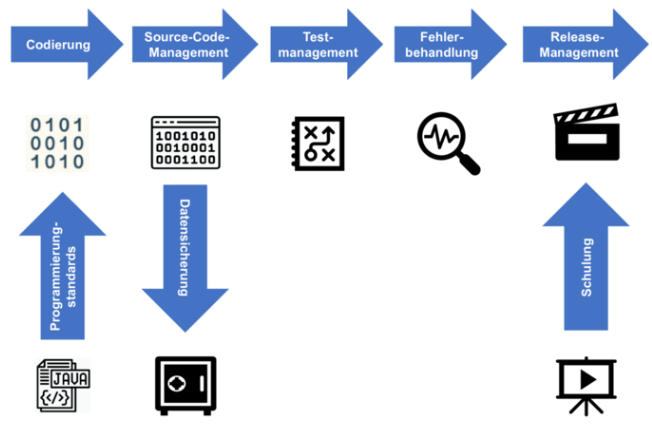
Abbildung 1 Entwicklungsmethodik
Der Prozess der Transformation von Anforderungen in die funktionalen und technischen Spezifikationen bis hinein in die Source-Code-Entwicklung ist in der Regel keine 1:1-Abbildung und daher relativ komplex. Um die n:n-Beziehungen darzustellen, soll folgende Beispiel betrachtet werden:
- Eine Benutzeranforderung lautet z. B. URS 1000: Auf den Datensätzen # 3, 8 und 9 soll eine Audit-Trail-Funktionalität vorhanden sein. Zudem sollen Änderungen der genannten Datensätze nur für die Anwendergruppe „Sachkundige Person“ möglich sein.
- Funktional würde der Lieferant mit den Funktionen „Audit-Trail“, „Rechteverwaltung“ und „Benutzerverwaltung“ antworten.
- Ein Entwickler würde dies nun modular in Entwicklungsaufgaben (Units) einteilen:
- Erstellung der Audit-Trail-Funktionalität (diese würde er nur einmal programmieren und den Datensätzen 3, 8 und 9 zuweisen)
- Verknüpfung der Audit-Trail-Funktion mit den Benutzerdaten
- Anpassung der Systemkonfiguration (Audit-Trails)
- generisches Rechtesystem verbunden mit Benutzerdaten und Datensätzen
- Eine weitere Benutzeranforderung könnte sein: URS 1010: Audit-Trail-Daten sollen für die Anwendergruppe „admin“ ausdruckbar sein.
- Nun würde der Entwickler nicht eine komplett neue Funktion erstellen, sondern die Audit-Trail-Funktionalität und Rechteverwaltung aus der ersten Anforderung/Umsetzung entsprechend erweitern (z. B. Druck-Button) oder modifizieren.
Dieses Beispiel zeigt, dass es eine n:n-Beziehung zwischen Anforderungen, Funktionen und Entwicklungsaufgaben gibt. Solche Beziehungen werden am Besten in einer Beziehungsmatrix dargestellt. Im einfachsten Fall reicht ein Tabellenkalkulationsprogramm, in komplexeren Fällen empfiehlt sich eine relationale Datenbank.
1 determination ersetzte evaluation, aber die Abkürzung blieb
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











