

Outsourcing – Aufgaben des Auftraggebers
Auszug aus dem GMP-BERATER, Kapitel 1.L.3, Aufgaben des Auftraggebers
8 Min. Lesezeit | von Dr. Frank Böttcher
Erschienen im LOGFILE Leitartikel 13/2022
Die Einhaltung der rechtlichen Vorgaben für die Herstellung und Prüfung eines Arzneimittels obliegt in Europa der Sachkundigen Person.
Das Management des Auftraggebers muss der Sachkundigen Person aber die notwendigen Ressourcen zur Verfügung stellen, damit sie ihrer arzneimittelrechtlichen Verantwortung nachkommen kann. Sie kann Aufgaben delegieren, insbesondere an den Leiter Herstellung und Leiter Qualitätskontrolle, oder auch an andere Sachkundige Personen, die im Unterauftrag tätig werden. Dennoch trägt die Sachkundige Person, die die Freigabe zum Inverkehrbringen des Arzneimittels bescheinigt, die Verantwortung dafür, dass das Arzneimittel nach den gesetzlichen Regelungen und in Übereinstimmung mit der Zulassung oder Genehmigung für die klinische Prüfung hergestellt und geprüft wird.
Die Behörden fragen zunehmend nicht nur nach der Qualifikation der Sachkundigen Person, sondern auch danach, ob sie die entsprechende Erfahrung mit den von ihr freizugebenden Produkten hat. Da die Anforderungen an die sehr unterschiedlichen Verfahren bei der Herstellung und Prüfung der verschiedenen Arzneimittel immer komplexer werden, kann es daher zukünftig notwendig werden, auch bei der Zertifizierung kritischer Prüfverfahren auf die Unterstützung der Sachkundigen Person der Dienstleister zurückzugreifen. Die Kompetenz der Sachkundigen Person der Labordienstleister wird dabei weniger für die Chargenfreigabe genutzt, sondern vielmehr für die Zertifizierung spezieller Prüfungen, für die bei der Sachkundigen Person des Auftraggebers keine ausreichenden Erfahrungen vorliegen. So kann die Expertise des Dienstleistungslabors genutzt werden, um die Sachkundige Person, die für die Marktfreigabe verantwortlich ist, besser zu unterstützen. Sie muss allerdings für den Auftraggeber trotz der Vergabe einzelner Leistungen an Unterauftragnehmer letztlich sicherstellen, dass alle arzneimittelrechtlichen Vorgaben eingehalten werden.
Der Auftraggeber muss in seinem QS-System genau beschreiben, wie die Sachkundige Person dieser Verantwortung gerecht werden kann. Darüber hinaus muss der Auftraggeber in seinem QS-System festlegen, wie die Qualifizierung des Zulieferers erfolgen soll und wie die Leistung des Zulieferers kontinuierlich überprüft werden kann.
Grundlage für die Zusammenarbeit ist der Abschluss eines Vertrages mit dem Auftragnehmer, der die Vergabe und Aufteilung der Verantwortung zwischen den beteiligten Unternehmen genau regelt (GMP-Vertrag bzw. Verantwortungsabgrenzungsvertrag).
Vor Vergabe der Aufträge an Zulieferer muss der Auftraggeber sich davon überzeugen, dass die Auftragsvergabe in Übereinstimmung mit den gesetzlichen Vorgaben erfolgt, der Auftragnehmer geeignet ist und die nötigen Einrichtungen, Kompetenzen und das Personal besitzt, um die Tätigkeit im Auftrag ohne Qualitätseinbußen zu übernehmen. Dazu gehört auch, dass der Auftraggeber die Einhaltung der GMP-Regularien beim Auftragnehmer sicherstellen muss.
Der Auftraggeber muss ferner dafür sorgen, dass der Auftragnehmer alle Informationen und Unterlagen von ihm erhält, die ihn in die Lage versetzen, die Tätigkeiten zulassungskonform auszuführen. Dazu ist es notwendig, nicht nur Herstellungs- und Prüfanweisungen auszutauschen, sondern den Auftragnehmer auch darüber zu informieren, welche Probleme bei der Entwicklung oder in der Vergangenheit bei der Herstellung und Prüfung aufgetreten sind. Daneben sind Unterlagen zu Validierungen oder Revalidierungen, ggf. Auswertungen aus dem Product Quality Review (PQR) für die schnelle und sichere Übertragung einer Leistung an einen Dienstleister von großer Bedeutung. Ferner muss der Auftraggeber alle Informationen zu eventuell notwendigen Sicherheitsvorkehrungen an den Auftragnehmer weitergeben. Dies ist natürlich bei Arzneimitteln und Wirkstoffen aufgrund ihrer pharmakologischen Aktivität zwingend notwendig, insbesondere bei noch nicht allgemein bekannten Substanzen aus Neuentwicklungen.
Im Rahmen der kontinuierlichen Lieferantenbewertung muss der Auftraggeber regelmäßig die Leistung seiner Zulieferer bewerten. Er muss außerdem sicherstellen, dass notwendige Änderungen und Ergänzungen bestehender Vereinbarungen oder Vorgaben erfolgen, wenn er feststellt, dass dies notwendig wird. Diese Bewertung kann risikobasiert erfolgen und sollte verschiedene Dinge berücksichtigen. Dazu gehört, ob die Leistung einmalig oder dauerhaft vergeben wird, welche Erfahrungen in der bisherigen Zusammenarbeit mit dem Zulieferer gemacht wurden, wie Reklamationen und Abweichungen bearbeitet wurden, die Bewertung der Ergebnisse eigener Audits oder die der Überwachungsbehörden.
Der Auftraggeber ist ferner dafür verantwortlich, die Dokumentation, die zu einer bestimmten Leistung gehört, zu sichten und zu bewerten. Dies kann direkt durch die Sachkundige Person des Auftraggebers erfolgen oder auch an eine Sachkundige Person des Auftragnehmers delegiert werden, wenn dies vertraglich festgelegt wurde. Auch hier ist ein risikobasiertes Vorgehen anzuraten, damit nicht einzelne Dokumente doppelt geprüft werden müssen. Die Prüfung sollte aber möglichst von der Sachkundigen Person mit der größten Expertise für den zu bewertenden Prozessschritt oder die zu bewertende Prüfung durchgeführt werden.
Ob die Dokumentation beim Auftragnehmer für den Auftraggeber ausreichend ist, kann dadurch verifiziert werden, dass bei erstmaliger Vergabe die Dokumente der ersten Chargen komplett dem Auftraggeber zur Verfügung gestellt werden. Später können im Rahmen regelmäßiger Lieferantenaudits stichprobenartige Überprüfungen erfolgen.
Im jährlichen Product Quality Review muss regelmäßig nachgewiesen werden, dass die Vereinbarungen zwischen Auftraggeber und Auftragnehmer aktuell sind. Neben der Überprüfung der Aktualität der Vereinbarungen zwischen Auftraggeber und Auftragnehmer und der Trendanalysen wird der Product Quality Review zukünftig eine größere Rolle im Prozessverständnis und für die Weiterentwicklung der Prozesse spielen, da hier die relevanten Daten zum Produkt, aber auch Informationen zu Verfahren selbst zusammengefasst und bewertet werden. Anhand dieser Informationen können auch die Prüfverfahren hinsichtlich Ihrer Eignung, notwendiger Revalidierungen oder Verbesserungen hin überprüft werden.
Die in Abbildung 1.L-4 aufgeführten Aufgaben des Auftraggebers umfassen dabei nur die formellen Anforderungen. Die Komplexität der eingesetzten Verfahren zur Prüfung von Produkten oder auch zur Steuerung der Herstellungsprozesse nimmt ständig zu. Damit steigt auch die Einbindung externer Experten wie Labordienstleister. Aus diesem Grund müssen Schnittstellen definiert werden, um das beim Dienstleister für die Produkte des Auftraggebers vorhandene und sich stetig weiter entwickelnde Know-how für das prozessorientierte Wissensmanagement beim Auftraggeber verfügbar zu haben. Dies gilt auch bei einem evtl. Wechsel des Dienstleisters. In diesem Fall ist es wichtig, das Wissen beim Transfer zu einem neuen Dienstleister zur Verfügung zu haben.
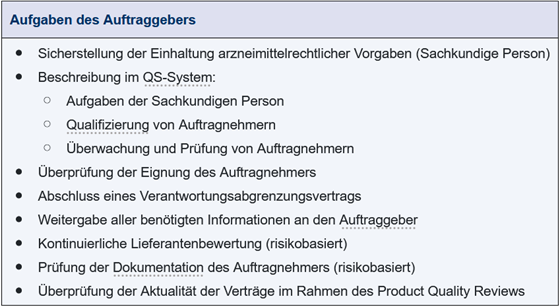
Abbildung 1.L-4 Aufgaben des Auftraggebers
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











