

Prüfintervall für die Integritätsprüfung von HEPA-Filtern in einem Laminar Flow
Ihre Frage – unsere Antwort
12 Min. Lesezeit | von Harald Flechl
Erschienen im LOGFILE Leitartikel 29/2021
Frage:
Wir betreiben in einem Reinraum, GMP-Reinraumklasse C (ISO 8 in operation, ISO 7 / at rest), ein Laminar Flow über einer Spritzgussmaschine.
Das Laminar Flow erreicht innerhalb des Reinluftbereichs ISO-Klasse 5.
Wie oft sollte die Prüfung auf Integrität des endständigen HEPA-Filters durchgeführt werden? Gibt es eine Vorgabe? Oder müssen wir eine Risikoanalyse durchführen und die Intervalle danach festlegen?
Antwort:
Zusammenfassende Empfehlung
A) Erfolgte die (gewerberechtliche) Betriebsgenehmigung nach rein gesetzlichen Anforderungen, ist eine dokumentierte Risikobeurteilung zur Begründung der Prüfintervalle auf alle Fälle zu empfehlen.
B) Ist die Betriebsgenehmigung zusätzlich zu den gesetzlichen Anforderungen auch nach EU-GMP-Richtlinien erfolgt, ist es bis zur Revision des Annex 1 ausreichend, eine Risikobeurteilung zur Begründung der Prüfintervalle dokumentiert durchzuführen (siehe unter „Welche Punkte sollte die Risikobetrachtung umfassen?“).
Seitens der Behörden wird jedoch für die ISO-Klasse 5 (Laminar-Flow wird mit GMP-Klasse A gleichgesetzt) eine halbjährliche Prüfung erwartet. In der Revision des Annex 1 (Entwurf 2020) sind diese Intervalle schon beschrieben (siehe unter „Welche GMP-Regularien und Normen greifen hier?“).
Empfohlenes Prüfintervall für die Prüfung auf Integrität der HEPA-Filter
- bei der Erstinstallation oder Austausch des Filters
- wiederkehrende Prüfung bei neuen Filtern nach einem Jahr, danach nach 3 Jahren, wenn nicht vorher ein anderes Ereignis wie in EN ISO 14644-3, Anhang A.3, a) bis d) auftritt (siehe Abschnitt „Welche GMP-Regularien und Normen greifen hier?“)
- bei Erreichen der vom Hersteller angegebenen Lebensdauer unter den gegebenen Betriebsbedingungen sollte das Intervall auf jährlich oder halbjährlich (Ergebnis aus der Risikobeurteilung) reduziert werden.
- bei Erkennen von Partikelverunreinigungen am Produkt, die durch luftgetragene Verunreinigung erfolgt.
Welche Punkte sollte die Risikobetrachtung umfassen?
Ein Hinweis zur periodischen Überprüfung könnte in der Betriebsanlagengenehmigung enthalten sein. Auch nach welchen gesetzlichen Vorgaben die Genehmigung erteilt wurde, sollte dort ersichtlich sein.
Wenn die Anlagen und die Produktion gemäß GMP genehmigt sind und betrieben werden, sind trotzdem in erster Linie die gesetzlichen Vorgaben maßgebend. Obwohl die GMP-Guidelines rechtlich als „Empfehlung“ gelten, werden die Empfehlungen der Guidelines von den Behörden/Inspektoren als „Vorgabe“ angesehen und auch so bewertet. Nachdem es sich um eine Spritzgussmaschine handelt kann je nach hergestelltem Produkt und der Verwendung (z. B. als „Zubehör eines Medizinprodukts“) auch die Medizinprodukte-Richtlinie maßgebend sein. Ergänzend dazu kann der EU-GMP-Leitfaden, wobei eine „aseptische“ Produktion ausgeschlossen werden kann, der Genehmigung zugrunde liegen, wobei die „nicht sterile Fertigung“ auch keine Klasse A (ISO 5) oder B festlegt. Der GMP-Leitfaden bezieht sich wiederum bei reinraumtechnischen Themen auf die ISO 14644-Normenreihe.
Das bedeutet: Wenn die Genehmigung unter dem EU-GMP-Leitfaden erteilt wurde, sollte man die „Vorgaben“ des GMP-Leitfadens ergänzend zur ISO 14644-Reihe für die Beurteilung heranziehen. Wenn das nicht der Fall ist, also eine rein „gewerberechtliche“ Betriebsgenehmigung vorliegt, kann man nur die ISO-Richtlinien als Beurteilungsgrundlage verwenden.
Alle Guidelines haben eines gemeinsam bei der Festlegung eines Prüfintervalls: das Risiko soll beurteilt werden.
Bei einer Risikobetrachtung zur Beurteilung eines Lecks in einem HEPA-Filter (Klasse H13 oder H14 nach EN 1822) wären folgende Punkte zu betrachten (kein Anspruch auf Vollständigkeit):
- Das Wichtigste: Welche Auswirkung hat ein potentielles Leck im Filter auf mein Produkt? D. h. welche Gefahr besteht für die Person, die dieses Produkt anwendet, bei einer möglichen Verunreinigung durch ein Leck im HEPA-Filter?
(Im Extremfall wäre das Ergebnis: Es wird keine Reinheitsklasse ISO 5 benötigt - damit sind die weiteren Punkte obsolet!) - Wie ist im Normalbetrieb die Verunreinigung der Luft unmittelbar vor dem HEPA-Filter? Sind Vorfilter eingesetzt, wenn ja, welche Abscheideleistung haben diese Filter? (Dient zur Beurteilung welche Auswirkung ein Leck im HEPA-Filter haben kann. Untersuchungen haben gezeigt, dass bei einem Leck ca. 3 % der Partikel vor dem HEPA-Filter durch das Leck penetrieren können.)
- Ein Leck in einem HEPA-Filter entsteht nicht von selbst, sondern meist durch mechanische Belastung/Beschädigung beim Hantieren mit dem Filter beim Einbau (wird erkannt bei der Abnahmemessung = Funktionsprüfung).
Besteht die Gefahr einer Filterbeschädigung bei Manipulation im Werkzeugbereich (Werkzeugtausch, Reinigung, o.ä.)? Wenn das der Fall sein kann, wäre lt. Risikobeurteilung eine Leckprüfung in diesem Fall anzuraten. - Nach einer gewissen Betriebszeit des Filters und abhängig von den Umgebungsparametern (Temperatur, Luftfeuchte) kann die Dichtmasse, mit der das Filterpaket in den Rahmen eingedichtet ist, „spröde“ und luftdurchlässig werden. Bei „normalen“ Temperaturen (ca. 19 °C bis 25 °C, ca. 35 % bis 65 % rel. Feuchte) kann eine Lebensdauer der Filter mit 5-10 Jahren angenommen werden - genaueres kann der Filterlieferant bestätigen (Shelf-Life). Erst ab dieser Zeit könnte man eventuell ein Leck detektieren (Anmerkung: Ich kenne HEPA-Filter in Klasse A und B die seit 20 Jahren in Betrieb sind.)
- Die Leckmessung (Integritätsprüfung) eines HEPA-Filters ist zwar keine Wissenschaft, aber der Messtechniker braucht entsprechende Erfahrung und das richtige Messequipment. Es werden ca. 80 % der erkannten Lecks durch Fehlmessungen festgestellt (Induktion; falsche Messanordnung; Sondendurchmesser ist nicht isokinetisch auf die tatsächliche Luftströmung abgestimmt; Lecktest wird als „Abscheidegradmessung“ durchgeführt; o.ä.).
- Innerhalb des „Laminar-Flow“-Bereiches sollte man die Luftströmungen kennen - Sichtbarmachung durch „Strömungsvisualisierung“, um festzulegen, in welchem Bereich ein Leck kritisch für das Produkt sein könnte bzw. ob der thermische Auftrieb zu einer Rückströmung im kritischen Bereich führt.
- Der thermische Auftrieb kann auch die Ursache für eine festgestellte Verunreinigung sein und hat dann mit der Abscheideleistung des Filters (Leck) nichts zu tun (vgl. Punkt 6).
- Eine Filterverschmutzung (Staubspeicherung) im Filter ist dem Zweck des Filters geschuldet und keine „Qualitätsverschlechterung“. Im Gegenteil, die Filterverschmutzung erhöht die Abscheideleistung des Filters etwas. Durch die Verschmutzung erhöht sich jedoch der luftseitige Druckverlust und ist dann ein energetisches Problem (der Anstieg bis zum Berstdruck des Filters wird meist durch die Ventilatoren nicht erreicht).
Bei einer „Laminar-Flow“-Anlage über einem Spritzgusswerkzeug mit hoher Temperatur ist die verbreitet angenommene „Richtgeschwindigkeit“ von 0,45 m/s (± 20 %) durch den entstehenden thermischen Auftrieb meist nicht ausreichend und wird oft mit 0,60 m/s oder höher angesetzt. Dabei werden auch die Filter nicht mit der „Nennluftmenge“ bei einer Anströmgeschwindigkeit über die Filternennfläche [z.B. 610 x 610 mm = 0,37 m²] von 0,45 m/s betrieben, sondern meist mit größerer Luftmenge betrieben. Bei einer höheren Luftmenge als der vom Filterhersteller angegebenen Nennluftmenge reduziert sich die Filtereffektivität (Abscheideleistung). Es könnte dann ein Filter der Klasse H14 nur mehr einer Filterklasse H13 entsprechen, was aber kaum Auswirkungen auf die Luftreinheit haben wird. Wichtig dabei: Die Partikelsonde für die Leckmessung ist dieser Luftgeschwindigkeit anzupassen (siehe Punkt 5).
Welche GMP-Regularien und Normen greifen hier?
Im aktuell gültigen Annex 1 (2008) des EU-GMP-Leitfadens wird die Überwachung der Reinräume und Reinluftanlagen wie folgt beschrieben:
8. Reinräume und Reinluftanlagen sollten routinemäßig im Betriebszustand überwacht werden. Die Überwachungspunkte sollten auf einer formalen Risikoanalyse und den Ergebnissen, die bei der Klassifizierung der Räume und/oder Reinluftanlagen erhalten wurden, basieren.
9. Klasse A-Bereiche (ISO 5) sollten während der gesamten Dauer kritischer Fertigung einschließlich des Einrüstens der Anlage überwacht werden, es sei denn, es wird nachgewiesen, dass im Prozessverlauf Verunreinigungen den Partikelzähler beschädigen könnten oder eine Gefährdung darstellen, z. B. durch lebende Organismen oder radioaktive Gefährdung. In solchen Fällen sollte die Überwachung während der Anlageneinrüstung durchgeführt werden, bevor dieses Risiko eintreten kann. Die Überwachung während simulierter Abläufe sollte ebenfalls durchgeführt werden.
Klasse A-Bereiche sollten so häufig und mit angemessener Probengröße überwacht werden, dass alle Eingriffe, kurzzeitigen Ereignisse und jede Verschlechterung des Systems erfasst werden und Alarm ausgelöst wird, wenn die Warngrenzen überschritten werden. Als Folge der Generation von Partikeln oder Tröpfchen durch das Produkt ist es anerkannt, dass es nicht immer möglich ist, einen niedrigen Level von ≥ 5.0 µm Partikeln nachzuweisen.
Im Entwurf des Annex 1 (2020) - wird erst voraussichtlich Ende 2021 veröffentlicht werden und in 2022 Gültigkeit erlangen - ist eine periodische Prüfung vorgesehen:
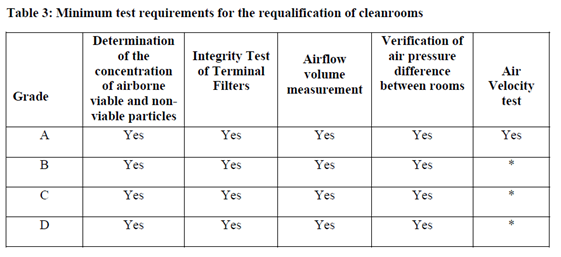
Intervalle für die Prüfung:
Für Klasse A (ISO 5) & B ist das maximale Intervall für die “Requalifizierung” 6 Monate, für Klasse C & D ist das maximale Intervall mit 12 Monaten vorgesehen.
Die EN ISO 14644-2:2016 beschreibt in Abschnitt 5:
5. Periodische Klassifizierung der Luftreinheit nach Partikelkonzentration
Die periodische Klassifizierungsprüfung muss jährlich gemäß ISO 14644-1 durchgeführt werden. Diese Häufigkeit kann auf der Grundlage der Risikobewertung, des Umfangs des Überwachungssystems und der Daten die durchgängig mit den im Überwachungsplan festgelegten Akzeptanzgrenzen oder -niveaus übereinstimmen.
Anmerkung: ISO 14644-3 spezifiziert ergänzende Tests, die sich auf andere Aspekte der Reinraumleistung beziehen, wie Druckdifferenz, Luftstrom, etc.
Die EN ISO 14644-3:2020 beschreibt im Anhang:
A.3 Planung von Prüfungen und Nachprüfungen Prüfungen sollten mindestens wie folgt durchgeführt werden:
a) in Verbindung mit der Klassifizierung nach ISO 14644-1;
b) bei der Nachprüfung während der Erst-Inbetriebnahme;
c) bei der Nachprüfung nach Erkennen und Beheben von Störungen;
d) bei der Nachprüfung nach einer Modifikation;
e) während der wiederkehrenden Prüfung.
Risikobeurteilungen sollten durchgeführt werden, um geeignete Intervalle für wiederkehrende Prüfungen festzulegen. Überwachungsdaten, Trends und Prüfergebnisse sollten zur Bestätigung und ggf. Anpassung der Zeitintervalle für die ausgewählten Prüfungen verwendet werden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











