

QRM in der Pharmaindustrie – Entwicklung einer Kontrollstrategie
Auszug aus dem GMP-BERATER, Kapitel 19.E.1, Konzeptionsphase
8 Min. Lesezeit | von Martin Mayer
Erschienen im LOGFILE 31/2022
Produktionsprozesse müssen kontrolliert ablaufen. Um dies zu erreichen, muss während der pharmazeutischen Entwicklung beziehungsweise während eines Technologietransfers eine wirksame Kontrollstrategie entwickelt werden. Dazu müssen die Prozessvariablen bekannt sein.
Diese können mit Hilfe von Prozessfließbildern identifiziert werden. Dann wird ermittelt, ob eine Prozessvariable als Prozessparameter oder Produkt-Qualitätsattribut angesehen werden muss:
- Prozessvariablen, die direkt gesteuert werden können und Einfluss auf ein Prozessergebnis haben, gelten als Prozessparameter.
- Prozessvariablen, die nicht direkt gesteuert werden können und das Ergebnis eines Prozessschrittes sind, gelten als Qualitätsattribut.
Zur Unterscheidung zwischen Prozessparametern und Qualitätsattributen kann ein Entscheidungsbaum genutzt werden, der entsprechend der Fragestellung gestaltet wird. Wie dies aussehen könnte, zeigt Abbildung 19.E-1.
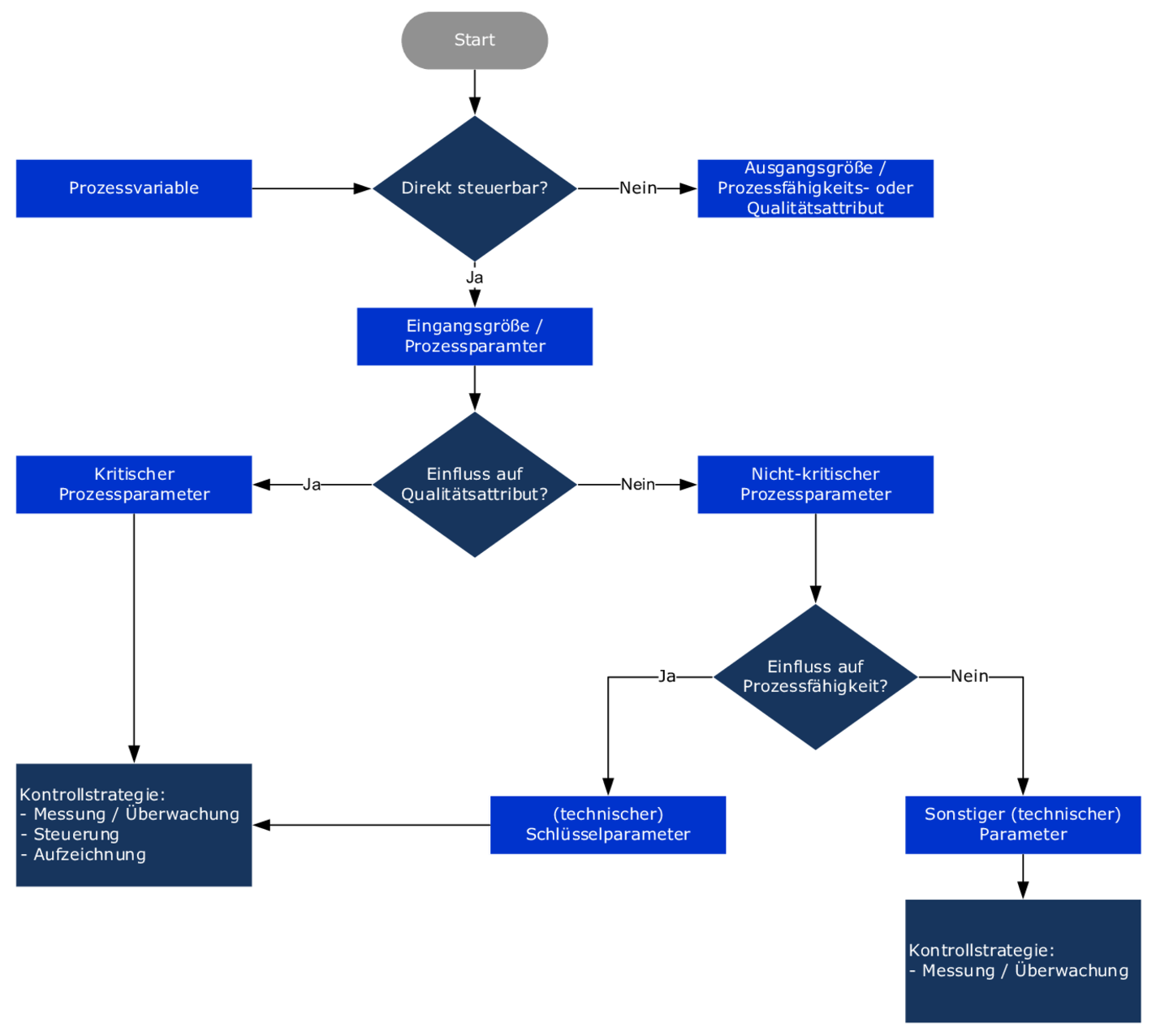
Abbildung 19.E-1 Entscheidungsbaum Kontrollstrategie, verändert nach PDA TR 60, Abb. 3.6-1
Um eine Kontrollstrategie festzulegen, sollte mittels QRM zwischen kritischen und nicht-kritischen Prozessparametern unterschieden werden. Prozessparameter, die Einfluss auf ein Produkt-Qualitätsattribut haben, sind als kritische Prozessparameter einzustufen. Das Ausmaß des Einflusses kann mittels QRM qualitativ, bzw. wenn möglich quantitativ bestimmt werden. Hierbei kommen Elemente einer FMEA zur Anwendung. Folgende Aspekte werden bewertet:
- Auftretenswahrscheinlichkeit von Abweichungen,
- deren Erkennbarkeit, bzw. Wahrscheinlichkeit der Nichterkennung,
- mögliches Ausmaß des Schadens im Falle einer Abweichung.
Kritische Prozessparameter sollten aktiv überwacht (gemessen und angezeigt) und (technisch) gesteuert werden; die Messwerte sind aufzuzeichnen.
Nicht-kritische Parameter können weiter untergliedert werden. Man unterscheidet zwischen (technischen) Schlüsselparametern und sonstigen (technischen) Prozessparametern:
- Parameter, die Einfluss auf die Prozessfähigkeit haben, werden als (technische) Schlüsselparameter klassifiziert. Auch für Schlüsselparameter sollte die Bewertung von Auftretenswahrscheinlichkeit, deren Erkennbarkeit, bzw. Wahrscheinlichkeit der Nichterkennung und dem möglichen Ausmaß des Schadens im Falle von Abweichungen erfolgen. Schlüsselparameter sollten ebenfalls gemessen (und angezeigt), gesteuert und aufgezeichnet werden.
- Parameter ohne Einfluss auf die Prozessfähigkeit gelten als sonstige (technische) Prozessparameter.

Abbildung 19.E-2 Wichtige Begriffe zur Entwicklung einer Kontrollstrategie
Die Messwerte kritischer Prozessparameter sollten in das Chargenprotokoll eingehen und zur Qualitätsbewertung durch die Sachkundige Person herangezogen werden. Für Schlüsselparameter kann die Information konform/nicht-konform im Chargenprotokoll ausreichend sein.
Wozu dient das Ergebnis des QRM-Prozesses?
Durch die intensive Durchleuchtung des Prozesses zur Identifizierung von (kritischen) Prozessparametern, Qualitätsattributen und (technischen) Schlüsselparametern wächst das Prozess- und Produktverständnis.
Die Identifizierung der kritischen Prozessparameter zeigt, wo die Kontrollstrategie ansetzen muss. Die Entscheidungen zur Kontrollstrategie sollten in das Design neuer Produktionsanlagen einfließen. Deren Umsetzung wird in der Qualifizierung überprüft. Sollen neue Prozesse auf bestehenden Anlagen gefahren werden, dient die Identifizierung der kritischen Prozessparameter zur Entscheidung, ob der neue Prozess durch die bestehende Qualifizierung abgedeckt ist.
Die Kenntnis der kritischen Prozessparameter und Qualitätsattribute bildet die Basis für die Prozessvalidierungsstrategie.
Die Messwerte von kritischen Parametern und von Schlüsselparametern sollten mittels statistischer Methoden ausgewertet und für Trendanalysen und Prozessfähigkeitsstudien genutzt werden.
Die Kontrollstrategie mit den Kontrollpunkten sollte in einem kontrollierten Dokument niedergeschrieben werden. Treten Abweichungen auf, kann zur Fehlerursachenanalyse eine Barriereanalyse genutzt werden; die Barrieren – die Kontrollpunkte – sind bekannt.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











