

Qualifizierung von Wasseranlagen – Ein Überblick
Auszug aus dem GMP:KnowHow Anlagenqualifizierung
7 Min. Lesezeit | von Fritz Röder
Erschienen im LOGFILE 21/2024
Reinstwassersysteme werden in pharmazeutischen Betrieben für eine Vielzahl von Anwendungen eingesetzt. Darunter fallen beispielsweise die Reinigung diverser Ausrüstungsgegenstände, die Reinstdampferzeugung sowie die Verwendung von Wasser als Ingredienz für feste, halbfeste und flüssige Arzneimittel. Aus diesem Grund hat eine Reinstwasseranlage einen hohen direkten und indirekten Einfluss auf die Qualität der Arzneimittel, was eine Qualifizierung unumgänglich macht.
Dass die Qualifizierung eines Wassersystems im Vergleich zu anderen Anlagen sehr aufwändig sein kann, ist technischen und natürlichen Prinzipien geschuldet. Daher ist eine Wasseraufbereitungsanlage auch sehr wartungsintensiv. Durch den Umstand, dass die Trinkwasserqualität natürlichen saisonalen Schwankungen unterliegt, dauert die Qualifizierungsphase einer neuen Anlage insgesamt nicht weniger als ein Jahr. Die Freigabe des Wassers für die Herstellung kann aber schon früher erfolgen.
Wie bei allen anderen Anlagen auch erfolgt die Qualifizierung formal nach dem bekannten Schema:
- Designqualifizierung (DQ)
- Installationsqualifizierung (IQ)
- Funktionsqualifizierung (OQ)
- Leistungsqualifizierung (PQ)
Die Grundlage für jede Qualifizierungsphase bilden eine Risikoanalyse sowie ein Qualifizierungsplan, in dem die Prüfpunkte spezifiziert werden. Aus dem genehmigten Plan wird ein Report (Bericht) erstellt und das Ergebnis der durchgeführten Tests dort eingetragen. Mit dem genehmigten Report ist die Qualifizierungsphase abgeschlossen.
In der Praxis lässt sich zunehmend eine „integrierte Qualifizierung“ beobachten, also die Verbindung von Commissioning und Qualifizierung. Das Konzept folgt ASTM E2500-13 und stellt die Subject Matter Experts, also die beteiligten Experten, in den Fokus. Resultat ist, dass doppelte Prüfungen im Rahmen von C&Q vermieden werden. Bereits beim Factory Acceptance Test (FAT) und Site Acceptance Test (SAT) können bestimmte Qualifizierungstests durchgeführt werden. Dieses Konzept ist auch bei Wasseranlagen anwendbar und sinnvoll.
Qualifizierung und Commissioning
Qualifiziert werden ausschließlich GMP-relevante Aspekte der Wasseranlage. Daneben gibt es auch nicht-GMP-relevante Aspekte, die aber trotzdem geprüft werden müssen, z. B. die Anforderungen der Maschinenrichtlinie. Das passiert im Rahmen des Commissioning (Inbetriebnahme). Wie aus der folgenden Abbildung ersichtlich ist, verlaufen Qualifizierung und Commissioning im Prinzip zeitlich parallel.
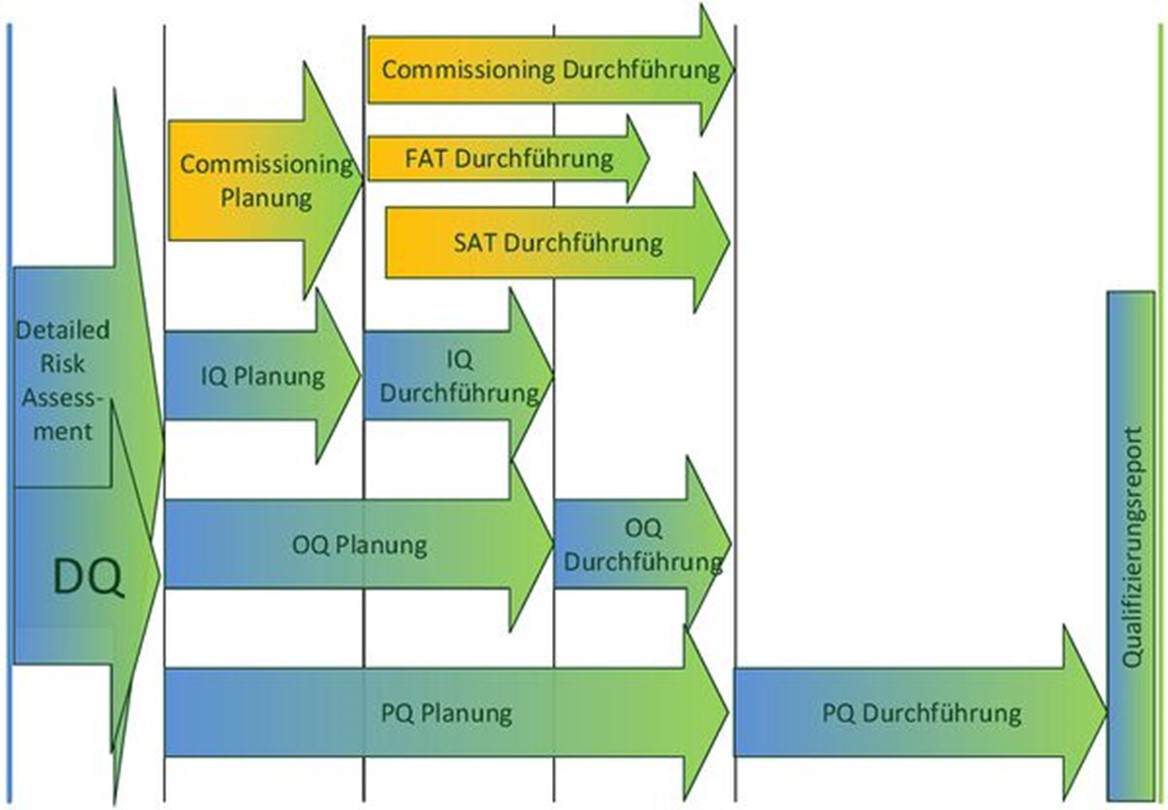
Betrachtet man den Zeitplan einer Wasseranlagen-Qualifizierung, so überschneidet sich die Planung und Durchführung der einzelnen Phasen grundsätzlich mit den nicht-GMP-relevanten Aktivitäten, die als Commissioning zusammengefasst werden. Beide Phasen überschneiden sich inhaltlich und zeitlich, und Tests werden ggf. übergreifend durchgeführt. Die „integrierte Qualifizierung“ kann im weiteren Verlauf reichlich Aufwand einsparen, setzt aber gutes Know-How voraus.
Terminplan und Zeitbedarf
Wer einen Terminplan für eine Qualifizierung aufsetzt, sollte grundsätzlich einen zeitlichen Puffer mit einplanen, und zwar aus folgenden Gründen:
- Während jeder Qualifizierung treten Probleme auf, die gelöst werden müssen. Das kann Zeit kosten.
- Auch wenn das Anlagendesign von Wasseraufbereitungsanlagen heute weitgehend standardisiert und reproduzierbar ist, treten in der Praxis immer Spezialfälle auf.
- Weiterhin erfordert der Abschluss der einzelnen Phasen in der Leistungsqualifizierung auch immer akzeptable Ergebnisse hinsichtlich der Wasserqualität. Am kritischsten ist dabei die Mikrobiologie zu betrachten. Bei der Terminplanung zu berücksichtigen ist hier die Inkubationsdauer der Proben, bis ein repräsentatives Ergebnis vorliegt.
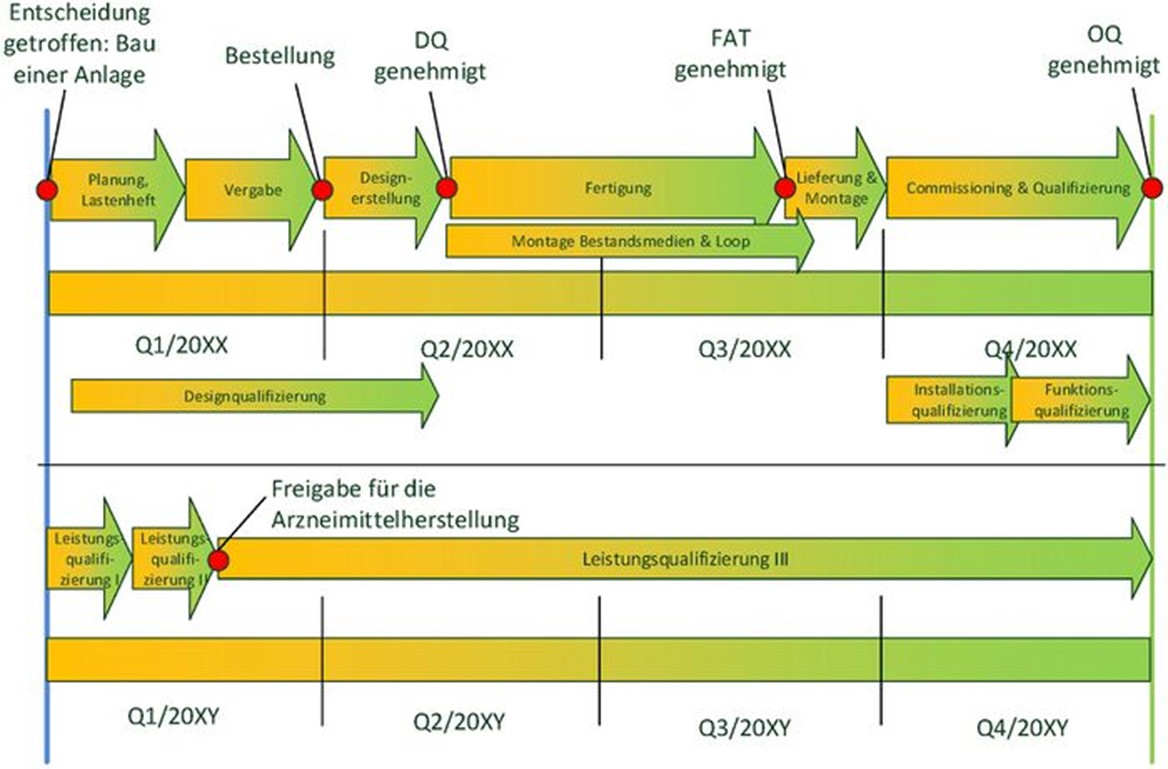
Die Performance Qualification ist typischerweise in drei Phasen unterteilt. Nach erfolgreichem Abschluss der ersten beiden Phasen kann das Wasser für die pharmazeutische Produktion verwendet werden. In Ausnahmefällen kann auch eine Freigabe des Wassers „at risk“ nach Phase eins erfolgen. Bei einer Überschreitung des Aktionslimits in Phase zwei müssen jedoch alle bis dahin hergestellten Produkte vernichtet werden.
In einem Projekt könnte der zeitliche Verlauf etwa so aussehen wie hier dargestellt:
Umfang der Qualifizierung
Bei der Qualifizierung von Wasseraufbereitungsanlagen muss betrachtet werden, welche Teile des Systems überhaupt neu gebaut werden. Dazu sollte man folgende Fragen stellen:
Wird ein Bestandssystem ersetzt?
In dem Fall kann man möglicherweise auf Bestandsdaten (Product Quality Review oder Trendbericht) zurückgreifen, um Limits und Spezifikationen zu definieren. Ist ein „neuer Standort“ geplant, an dem bisher kein Wasseraufbereitungssystem existiert hat, muss man auf Berechnungsformeln vertrauen. Eventuell gibt es in der Nähe Reinstwasseranlagen, deren Daten für Vergleichszwecke herangezogen werden können (Voraussetzung: gleiches Speisewasser).
Welche Teile der Reinstwasseranlage werden ersetzt?
Ein Verteilloop hat häufig eine längere Lebensdauer als eine Erzeugungseinheit. Dadurch kommt es häufig vor, dass die Systeme getrennt voneinander erneuert werden. Qualifiziert wird der Teil, der erneuert bzw. verändert wird. Allerdings hat auch die Erneuerung des einen Teils der Anlage Auswirkungen auf den anderen Teil. Eine genaue Betrachtung (Risikoabschätzung) im Vorfeld kann den Qualifizierungsaufwand reduzieren.Sind alle Fragen geklärt, können die Dokumente erstellt werden. Bei größeren Anlagenprojekten ist es gefordert, einen übergeordneten Masterqualifizierungsplan sowie eine Traceability Matrix anzulegen, um einen besseren Überblick zu behalten. Außerdem muss der Lieferant (rechtzeitig) qualifiziert sein.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











