

Qualitätsmerkmale guter GMP-Dokumentation
Auszug aus dem GMP-Fachwissen Papierbasierte und elektronische Dokumentation im Pharmaunternehmen, Kapitel 1.4.1
9 Min. Lesezeit | von Dr. Michael Hiob
Erschienen im LOGFILE Leitartikel 34/2022
„Dokumenteninhalte müssen ausreichend präzise sein, um den gewünschten Gebrauch sicherzustellen. Unschärfen im Dokument sind daher zu vermeiden. Begriffe wie „im Allgemeinen“, „meistens“, „in der Regel“ sind Indikatoren für Unschärfen in der Formulierung“, erläutert Regierungspharmaziedirektor Michael Hiob im GMP-Fachwissen „Papierbasierte und elektronische Dokumentation im Pharmaunternehmen“. Das E-Book zeigt Ihnen das 1x1 der guten Dokumentationspraxis im täglichen Umgang mit Dokumenten.
Welche weiteren formalen Anforderungen an GMP-Dokumente gestellt werden, lesen Sie heute in einem Auszug aus dem E-Book.
Sind Art und Umfang der in einem Pharmabetrieb verwendeten Dokumente auch recht unterschiedlich, so lassen sich doch für alle Dokumente allgemein geltende formale Anforderungen aufstellen, die bei der Erstellung zu beachten sind und folgende Aspekte betreffen:
- Anwendungsbereich und Ziele
- Schriftform
- Richtigkeit
- Vollständigkeit
- Genauigkeit
- Aktualität
- Verfügbarkeit
- Genehmigung
Anwendungsbereich und Ziele des Dokuments müssen erkennbar sein. Sie bestimmen wann, wo und wozu das Dokument anzuwenden ist. Werden Anwendungsbereich und Ziel eines Dokuments nicht klar formuliert, so bleibt Raum für Eigeninterpretationen des Lesers. Damit besteht die Gefahr, dass das Dokument falsch angewandt wird.
Anwendungsbereich und Ziel eines Dokuments müssen immer im Kontext der mitgeltenden Dokumente geregelt werden. Der Anwendungsbereich von Dokumenten sollte so breit wie möglich gehalten werden, um eine flexible Anwendbarkeit zu ermöglichen. Allgemeine Regelungen, wie der Umgang mit Prüfmitteln oder die Art der Dokumentation werden sinnvoller übergreifend und einheitlich für einen größeren Anwendungsbereich festgelegt.
Es gilt den Grundsatz zu verfolgen: Was allgemein geregelt werden kann, sollte auch in allgemein gültigen Dokumenten festgelegt werden. Nur spezielle Gegebenheiten erfordern auch spezielle Regelungen. Die Wiederholung allgemein geltender Verfahrensweisen in speziellen Anweisungen erhöht unnötig die Dokumentenmenge, verschlechtert den Überblick und erschwert die Überarbeitung der Dokumentation.
Schriftform: Der Grundsatz der Schriftform gilt sowohl für die Festlegung von Anforderungen in Anweisungen als auch für die Aufzeichnung von durchgeführten Tätigkeiten in Protokollen. Ob Aufzeichnungen auf Papier oder in elektronischen Datenverarbeitungssystemen erstellt werden, ist nicht erheblich. Bis auf das Eintragen von Daten sollen Dokumente jedoch nicht handgeschrieben sein (Anforderung des EU-GMP-Leitfadens, Kapitel 4.6). Handgeschriebene Eintragungen sollten gut lesbar und unauslöschbar (kein Bleistift!) sein. Maßgeblich ist, dass die relevanten Daten/Informationen tatsächlich vorhanden sind und lesbar gemacht werden können.
Richtigkeit: Die Dokumentation soll richtig sein. Ihr Inhalt soll, soweit zutreffend, mit den Zulassungsunterlagen, der Herstellungserlaubnis sowie sonstigen übergeordneten Dokumenten und den tatsächlichen Gegebenheiten vor Ort übereinstimmen. Die Überprüfung kritischer Daten wird üblicherweise nach dem Vier-Augen-Prinzip, d. h. mittels Gegenkontrolle durch eine zweite Person, oder mittels validierter elektronischer Verfahren vorgenommen.
Vollständigkeit: Alle Anweisungen und Aufzeichnungen müssen in lückenloser Form angefertigt und aufbewahrt werden. Die Vollständigkeit eines Dokuments sollte leicht erkennbar und überprüfbar sein, z. B. durch Angabe der Gesamtseitenzahl und durch ein Inhaltsverzeichnis.
Genauigkeit: Dokumenteninhalte müssen ausreichend präzise sein, um den gewünschten Gebrauch sicherzustellen. Unschärfen im Dokument sind daher zu vermeiden. Begriffe wie „im Allgemeinen“, „meistens“, „in der Regel“ sind Indikatoren für Unschärfen in der Formulierung.
|
"Desinfektionsmittelverdünnung aus Konzentraten werden nach den üblichen Verfahren hergestellt (außer Des-Infekt-Liquid). Die Verdünnung muss ausreichend lang einwirken, damit eine Keimreduktion eintritt. Wenn Desinfektionsmittel verdünnt wurden, sollten diese Lösungen nach Möglichkeit nicht zu lange aufbewahrt werden, weil es sonst zu Keimwachstum kommen könnte, wodurch man genau den gegenteiligen Effekt erzielen würde." |
Abbildung 1 Negativbeispiel für eine Formulierung aus einem Hygieneplan
Die in Abbildung 1 gezeigte Anweisung ist unpräzise: Was sind die „üblichen Verfahren“, wie lang ist „ausreichend lang“ oder „nicht zu lange“? Diese Anweisung ist auch mehrdeutig: Was heißt „außer Des-Infekt-Liquid“, „sollten“ oder „nach Möglichkeit“? Erläuterungen zum Keimwachstum, die dem Hintergrundwissen dienen, gehören in Schulungsveranstaltungen. In Anweisungen ist darzustellen, was zu tun ist, nicht warum etwas zu tun ist.
Abbildung 2 enthält eine Auswahl unpräziser Formulierungen, die oft in Anweisungen zu Unklarheiten und Mehrdeutigkeiten führen und zeigt, wie diese genauer formuliert werden könnten.
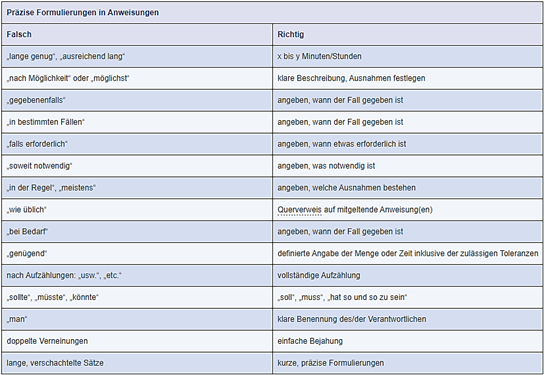
Abbildung 2 Präzise Formulierungen in Anweisungen
Dokumente sollen keine widersprüchlichen Aussagen enthalten, sondern konsistent sein. Dokumente sind insbesondere dann konsistent, wenn ihre Inhalte aufeinander abgestimmte Ziele und Realisierungswege verfolgen und die Beschreibung eines Dokuments mit jeweils geltenden anderen Dokumenten übereinstimmt. Die Anforderungen müssen den fachlichen und technischen Gegebenheiten entsprechen.
Der Inhalt von Dokumenten soll verständlich und nachvollziehbar sein, nicht nur für die Mitarbeitenden, die täglich mit den Dokumenten umgehen, sondern auch für Außenstehende, wie Inspektoren oder Auftraggeber. Der Inhalt des Dokuments soll eindeutig sein; Titel, Art und Zweck sollten klar bezeichnet sein.
Die Sprache des Dokuments ist das Medium, über das der Verfasser des Dokuments (Sender) mit dem Addressaten (Empfänger) kommuniziert. Die richtige Verwendung der Sprache entscheidet darüber, ob und wie der Inhalt des Dokuments beim Empfänger „ankommt“. Natürlich ist dies nicht allein eine Frage, ob z. B. ein Dokument in Deutsch, Englisch oder Französisch erstellt wird, obwohl oft in multinationalen Konzernen die Anweisungen der Muttergesellschaft im Original übernommen werden und von den Mitarbeitenden erwartet wird, dass sie alle Nuancen der Fremdsprache beherrschen, wie z. B. die englischen Unterscheidungen in „must“, „shall“ „have to“ oder „should“.
Die Sprache in Dokumenten ist vor allem eine Frage der Ausdrucksweise, d. h. in welchem Stil das Dokument geschrieben wird. Die Auswahl des richtigen Stils hängt u. a. von dem Addressaten und dem Anwendungsbereich ab. Dokumente sind verständlich, wenn sachkundige Leser den Inhalt des Dokuments unter Berücksichtigung seines Zwecks verstehen können. Dies bedeutet, dass die mit dem Thema befassten Personen z. B. auf Auftraggeber- und Auftragnehmerseite die Inhalte verstehen müssen.
So wird man eine Anweisung für Aushilfspersonal anders formulieren müssen als für ausgebildetes Fachpersonal, bei dem die Kenntnis von Fachbegriffen und Sachzusammenhängen vorausgesetzt werden kann.
In übergeordneten Dokumenten, die firmenstrategische Gesichtspunkte betrachten, wie z. B. Qualitätsmanagementleitlinien, werden Ziele formuliert. Die sprachlichen Formulierungen werden dementsprechend allgemein gehalten sein. Auf der operativen Ebene sind neben der Darstellung des Regelungsziels auch detaillierte Vorgaben erforderlich, wie das Ziel zu erreichen ist, beispielsweise bei einer Herstellungsanweisung. Diese Anweisungen müssen daher präzise und eindeutig sein.
Aktualität: Alle Aufzeichnungen müssen auf dem neuesten Stand sein. Dazu müssen Dokumente rechtzeitig erstellt und bei Bedarf an veränderte Gegebenheiten angepasst werden. Auch Protokolle und sonstige Aufzeichnungen müssen aktuell sein und müssen daher zum Zeitpunkt des jeweiligen Vorgangs angefertigt werden.
Verfügbarkeit: Die relevanten Dokumente müssen den Verantwortlichen vor Ort zur Verfügung stehen und benutzbar sein. Dafür müssen die Verantwortlichen das Dokument kennen, verstehen und anwenden können. Sie müssen über Änderungen des Dokuments rechtzeitig informiert werden. Wenn Aufzeichnungen mit elektronischen Datenverarbeitungssystemen erstellt werden, soll innerhalb angemessener Frist eine lesbare Schriftform hergestellt werden können.
Genehmigung: Dokumente müssen vor ihrer Verwendung durch die dafür Verantwortlichen genehmigt werden. Die Genehmigung setzt eine Überprüfung der Geeignetheit des Dokuments (Richtigkeit, Aktualität, Zweckmäßigkeit) voraus.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











