

Reinräume: Monitoring raumlufttechnischer Anlagen
Ein Auszug aus dem GMP-BERATER, Kapitel 3.K.3, Raumluft- und reinraumtechnische Daten
5 Min. Lesezeit | von Ing. Harald Flechl
Erschienen im LOGFILE 46/2022
Als Grundlage für das reinraumtechnische Monitoring können folgende Normen herangezogen werden:
- EN ISO 14644-2 Überwachung zum Nachweis der Reinraumleistung bezüglich Luftreinheit anhand der Partikelkonzentration
- ISO 14644-3 Prüfverfahren (für die Messtechnik)
- VDI 2083 Blatt 3.1 Messtechnik in der Reinraumluft – Monitoring
Nachstehend soll nur noch auf diejenigen Daten eingegangen werden, die im Rahmen des Pharmamonitorings der raumlufttechnischen Anlage zu belegen sind. Man muss dabei zwischen zwei Datentypen unterscheiden (siehe Abbildung 1).
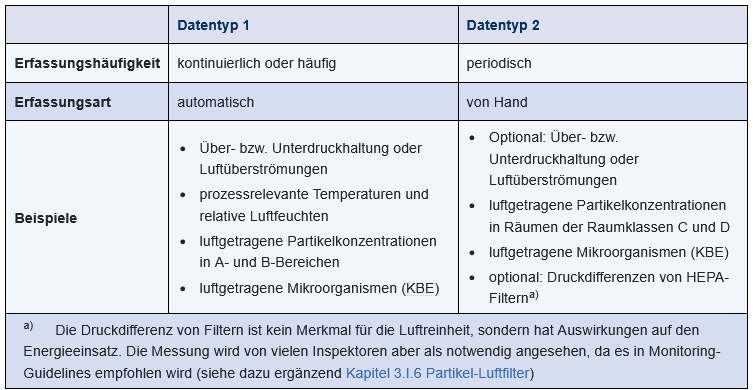
Abbildung 1 Datentypen für das Pharmamonitoring

Ist eine aktive Raumdruckregelung installiert und wird die Druckdifferenz zwischen Reinraum und Schleuse überwacht, dann sind die Raumdruck-Regelung und auch die Raumdruck-Alarmierung während der Türöffnung auszusetzen. Grund: das Öffnen der Schleusentüre hat nur einen sehr geringen Einfluss auf die Luftströmung zwischen Reinraum und Schleuse (die Strömung ist abhängig von den Luftmengenbilanzen aus der Differenz zwischen Zuluft und Abluft der verbundenen Räume), aber einen hohen Einfluss auf den Differenzdruck (der Druck gleicht sich aus und zeigt den Wert „0“). Störend ist das Öffnen von Flügeltüren – dabei wird eine Sog-/Stoßwirkung der Luft durch den Türflügel erzeugt, der unkontrollierte Luftströmungen verursacht.
Um die Frequenz, d. h. die Häufigkeit, der Datenerfassung zu bestimmen, ist eine risikobasierte Herangehensweise empfohlen und in der CCS zu begründen. Eine Ausgewogenheit zwischen einem effizienten Ressourceneinsatz und einem aussagekräftigen Gesamtbild der Daten ist anzustreben. Eine Beurteilung zur Ermittlung der Häufigkeit könnte beispielsweise folgende Faktoren in Betracht ziehen:
- Höhere Frequenz für Bereiche mit hoher Kontaminationswahrscheinlichkeit, z. B. Bereiche
- in der Nähe von kritischen „reineren“ Tätigkeiten
- mit höherem Aktivitätsgrad der Personen (ausgenommen Schleusen)
- mit höherem Materialfluss
- mit länger andauernden Aktivitäten
- mit exponiertem Produkt oder Materialien, die direkten Produktkontakt haben
- für die finale Formulierung
- für die Abfüllung
- deren Trendanalyse einen Anstieg der Partikelkonzentration zeigt (➜ CAPA erforderlich)
- Niedrigere Frequenz für Bereiche mit geringer Kontaminationswahrscheinlichkeit, z. B. Bereiche
- für „unreinere“ Tätigkeiten
- in denen kurzzeitige Öffnungen an Behältern mit kleiner Fläche der Öffnung erfolgen
- für geschlossene Prozesse
- deren Trendanalyse einen Abfall der Partikelkonzentration zeigt
Zu überwachen sind neben den Partikelkonzentrationen und den als kritisch definierten Druckdifferenzen alle mikrobiologischen Messgrößen, deren Erfassung und Auswertung insbesondere in der Herstellung steriler Arzneimittel ein besonders hoher Stellenwert zukommt, sie kann aber auch in der nicht-sterilen Herstellung erforderlich sein. In der FDA-Leitlinie für die aseptische Arzneimittelherstellung ist nur die Erfassung der luftgetragenen Mikroorganismen enthalten (aktive Messung), ergänzt durch die Luftkeimsedimentation (passive Messung) als optionale Messung.
Die Überwachung dieser Messgrößen wird auch in der Monographie <1116> der U.S. Pharmacopeia für die mikrobiologische Überwachung reiner Arbeitsbereiche empfohlen. Die numerischen Grenzwerte in dieser Monographie sind mit denjenigen im Anhang 1 zum EU-GMP-Leitfaden jedoch nicht deckungsgleich. Der Rhythmus der mikrobiologischen Datenerfassung ist risikoabhängig und kann von mehrmals pro Charge bis zu viertel- und halbjährlich reichen.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











