

Stabilität im neuen Gewand: Der ICH-Q1-Leitlinienentwurf
Auszug aus dem GMP-BERATER, Kapitel Aktuelle Themen
5 Min. Lesezeit | von Dr. Joachim Ermer
Erschienen im LOGFILE 16/2025
Welche Änderungen bei der Planung und Durchführung von Stabilitätsuntersuchungen an Arzneimitteln und Wirkstoffen sieht der Entwurf der ICH Q1-Leitlinie vor?
Dr. Joachim Ermer fasst die wichtigsten Änderungen zusammen und kommentiert kritische Aspekte.
Die ausführliche Erörterung aller geplanten Änderungen und Neuerungen finden Sie im GMP-BERATER in der Rubrik „Aktuelle Themen“.
Stabilitätsuntersuchungen sind ein essenzieller Bestandteil der Arzneimittelentwicklung und -zulassung. Sie dienen dazu, die Qualität, Sicherheit und Wirksamkeit eines Arzneimittels über einen definierten Zeitraum, die Verwendbarkeitsfrist, zu gewährleisten. Die Bedeutung wird auch dadurch verdeutlicht, dass die Stabilitätsleitlinie Q1A Stability Testing of New Drug Substances and Products das erste Thema war, welches im internationalen Harmonisierungsprozess aufgegriffen wurde (Oktober 1993).
Auf diese erste (Kern-) Leitlinie folgten weitere Aspekte von Stabilitätsprüfungen:
- ICH Q5C: Quality of biotechnological products: Stability Testing (Nov. 1995)
- ICH Q1B: Photostability Testing of New Drug Substances and Products (Nov. 1996)
- ICH Q1C: Requirements for New Dosage Forms (Nov. 1996)
- ICH Q1D: Bracketing and Matrixing Designs (Feb. 2002)
- ICH Q1E: Evaluation of Stability Data (Feb. 2003)
- ICH Q1F: Stability Data in Climatic Zones III and IV (Juni 2006, zurückgezogen)
In den 2000er-Jahren kam es zu wesentlichen Entwicklungen, zunächst in Bereich der pharmazeutischen Herstellung mit der ICH-Leitlinie Q8 zur pharmazeutischen Entwicklung (insbesondere die Anwendung von Quality-by-Design (QbD)-Prinzipien und -Werkzeugen) sowie der ICH-Leitlinie Q9 zum Qualitätsrisikomanagement. Der Fokus richtete sich auf eine holistische Betrachtung des gesamten Lebenszyklus, was zu den FDA- und EU-Leitlinien zur Prozessvalidierung führte. Diese Prinzipien fanden dann auch ihren Niederschlag in den weiteren ICH-Leitlinien, zum Beispiel Q12 zum Lebenszyklus-Management (hier zum ersten Mal auch für die Analytik und Qualitätskontrolle) sowie in der Revision der Validierungsleitlinie Q2 und der Entwicklung einer neuen ICH-Leitlinie Q14 zur analytischen Methodenentwicklung.
Konsequenterweise wurde dann 2022 die Entscheidung getroffen, die ICH-Stabilitätsleitlinien zu revisionieren. Neben der Berücksichtigung der zuvor genannten Prinzipien war ein wesentliches Ziel die Zusammenfassung der verschiedenen Leitlinien sowie der Arzneimittel mit chemischen und biologischen Wirkstoffen. Trotz der komplexen Aufgabenstellung, die durch die deutliche Erweiterung des ICH-Gültigkeitsbereichs sicher nicht leichter geworden ist, hat es die ICH-Expertengruppe geschafft, den 2022 aufgestellten Zeitplan einzuhalten, so dass der Leitlinien-Entwurf im April 2025 zur öffentlichen Konsultation veröffentlicht wurde. (https://www.ich.org/page/quality-guidelines)
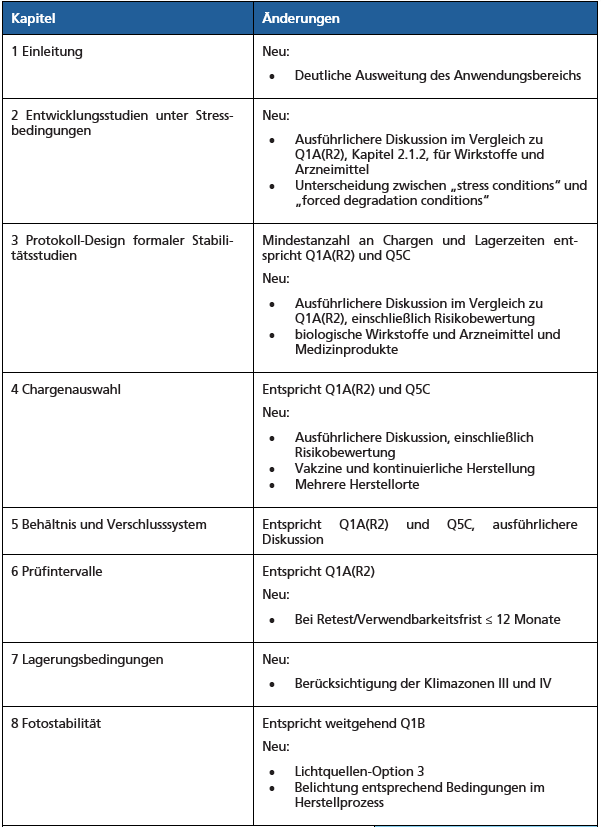
Im Folgenden erläutert der Autor aus seiner Sicht wichtige Ergänzungen, sowie einige Defizite und Verbesserungspotenziale des ICH Q1-Leitlinienentwurfs. Tabelle 1 zeigt eine Übersicht der wesentlichen Änderungen und Bezüge des Leitlinienentwurfs.
Tabelle 1: Übersicht der wesentlichen Änderungen in ICH Q1

Zusammenfassung
Die Anwendung der ICH Q9 Prinzipien des Risikomanagements bei allen Entscheidungen zieht sich durch den gesamten Q1-Leitlinienentwurf und wird für die entsprechenden Begründungen erwartet. Die Leitlinie wurde auch konsequent am Lebenszyklusansatz ausgerichtet, weshalb explizit gefordert ist, sie in ihrer Gesamtheit zu betrachten.
Für die folgenden Aspekte erlaubt der Entwurf eine größere Flexibilität als die Vorläuferleitlinien:
- Reduzierte Lagerzeiten, falls keine Änderung der CQAs oder Alternativen (Modellierung) vorliegen (Kapitel 6)
- Fotostabilität: Option 3: Leuchtstoff- oder LED-Lampe mit Umgebungslichtbedingungen (> 400 nm) während Herstellung, Verarbeitung und Anwendung
- Extrapolation für gefroren gelagerte synthetische Stoffe (Kapitel 13.2.8)
- Extrapolation für gefroren gelagerte biologische Stoffe (Kapitel 13.2.9)
- Anhang 1: Reduzierte Stabilitätsprotokolle
Einige Aspekte bedürfen nach Verständnis des Autors weiterer Diskussion oder sollten durch Beispiele zur Verdeutlichung ergänzt werden:
- Statistische Analyse:
Anwendung des Toleranzbereichs-Ansatzes (breiter als Vertrauensbereiche) sowie nichtlineare Regression - Höhere Stabilitätsmodellierungen:
Die Anmerkung „Nicht als Ersatz für Langzeit-Stabilitätsstudien gedacht“ steht im Widerspruch zu anderen Diskussionen („Eine stärkere Extrapolation kann mit anderen Modellierungsmethoden möglich sein (Anhang 2 – Stabilitätsmodellierung)“) - Entscheidungsbaum:
„Die Daten zeigen geringe oder keine Veränderung und geringe oder keine Variabilität.“ Wie viel ist „gering“ oder „nicht vorhanden“? „Keine Variabilität“ ist wissenschaftlich nicht fundiert!
Die folgenden Punkte sollten aus Sicht des Autors korrigiert bzw. ergänzt werden:
- Statistische Analyse:
Für CQAs mit oberen und unteren Akzeptanzkriterien und bekannter Änderungsrichtung (Vorwissen) ist die einseitige Vertrauensbereichsgrenze wissenschaftlich gerechtfertigt.
Die statistische Analyse sollte nur bei signifikanter Steigung durchgeführt werden, da eine Extrapolation der Vertrauensbereiche nur sinnvoll ist, wenn eine (wahre) Änderung vorliegt. - Entscheidungsbaum Szenario B:
Eine signifikante Änderung unter beschleunigten Bedingungen ist nicht zulässig. - Extrapolation:
Eine Extrapolation für gefroren gelagerte synthetische Stoffe ist bei keiner oder geringfügiger Änderung wissenschaftlich gerechtfertigt und sollte im Entscheidungsbaum berücksichtigt werden.
Fazit des Autors
Unter Berücksichtigung der komplexen Thematik und der inzwischen zahlreichen ICH-Stakeholder ist eine gute Balance zwischen Kontinuität, Weiterentwicklung und Modernisierung gelungen. Der Bedarf an Korrekturen und Ergänzungen ist sehr überschaubar. Ich möchte Sie jedoch ermutigen, aus Ihrer Sicht wichtige Aspekte im Kommentierungsprozess zu adressieren, um die Praxistauglichkeit der Q1-Leitlinie weiter zu erhöhen.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











