

Validierung analytischer Verfahren: Regulatorische Anforderungen – Teil 1
Auszug aus dem GMP-BERATER, Kapitel 14.D Validierung analytischer Verfahren
3 Min. Lesezeit | von Joachim Ermer
Erschienen im LOGFILE 16/2024
Analytische Verfahren werden überall in der Entwicklung, Herstellung und Freigabe von Arzneistoffen und -formen eingesetzt. Richtigkeit und Zuverlässigkeit von Analysenergebnissen sind deshalb essenziell. Die erfolgreiche Methodenvalidierung beweist dabei, dass das Prüfverfahren für den definierten Einsatz geeignet ist.
In unserem heutigen Leitartikel schreibt Joachim Ermer über die regulatorischen Anforderungen an die Validierung analytischer Verfahren.
Im nächsten LOGFILE erfahren Sie dann mehr zu wichtigen Aspekten bei der Umsetzung dieser Anforderungen in die Praxis.
Der Nachweis der Eignung analytischer Verfahren ist eine allgemeine GMP-Anforderung. Während der EU-GMP-Leitfaden Teil 1 recht allgemein „validierte Testmethoden“ fordert, verweist der Teil 2 hinsichtlich der Inhalte der Methodenvalidierung auf die ICH-Leitlinien. Auch der 21 CFR 211 fordert in §194(a)(2), dass die Eignung der Prüfmethoden verifiziert werden muss.
Die 1994 veröffentlichte ICH-Guideline Q2 A zur Methodenvalidierung war sehr wichtig für die Standardisierung der Begriffe und Definitionen sowie für die Festlegung der grundsätzlichen Anforderungen an die analytische Validierung. Die Leitlinie wurde 1995 von der EU als Scientific Guideline in die Notice to Applicants (EudraLex Volume 3) übernommen und ist damit verbindlich für Neuzulassungen. Im Jahr 2005 wurden die Leitlinien ICH Q2 A und Q2 B unter der Bezeichnung Q2(R1) mit dem neuen Titel Validierung von Prüfverfahren: Text und Methodologie zusammengeführt, ohne inhaltliche Änderungen.
Seit den neunziger Jahren gab es wesentliche Weiterentwicklungen, insbesondere im Bereich der pharmazeutischen Produktion, wie beispielsweise die Quality-by-Design Werkzeuge, Risikoanalyse, oder Lebenszyklus. Die Validierungsguideline Q2(R1) fokussiert sich im Wesentlichen auf chromatographische Prüfverfahren, auf Grund des Fehlens von Akzeptanzkriterien ist die Basis der geforderten Eignung diffus und das Kapitel „Linearität“ verwirrt die Responsefunktion (Kalibriermodell) mit der Linearität des Analyten in der Probe, d. h. mit der Richtigkeit. Letzteres führte zu Problemen bei Analysetechniken mit nichtlinearen Responsefunktionen, insbesondere bei biologischen Methoden und Produkten.
Deshalb wurde im November 2018 von der ICH ein Konzeptpapier zur Revision der Validierungsguideline und zur Einführung einer neuen Guideline Q14 zur Analytischen Methodenentwicklung veröffentlicht. Nach fast zwei Jahren Verzögerung wurden im März 2022 die Entwürfe der beiden Guidelines im Schritt 2 zur öffentlichen Konsultation veröffentlicht und erhielten jeweils mehr als 3000 Kommentare. Auf dem ICH-Meeting in Prag am 31. Oktober und 1. November 2023 wurden die finalisierten Guidelines im Schritt 4 von den regulatorischen ICH-Mitgliedern angenommen, allerdings erst am 20. Dezember 2023 auf der ICH-Webseite veröffentlicht (gültig seit 14. Juni 2024).
Einige der Defizite wurden bereinigt, aber erwartungsgemäß führte der internationale Abstimmungsprozess auch zu verschiedenen Kompromissen und Inkonsistenzen. Die – aus Sicht des Autors – wesentlichen Änderungen und Unzulänglichkeiten sind in folgender Tabelle zusammengefasst.
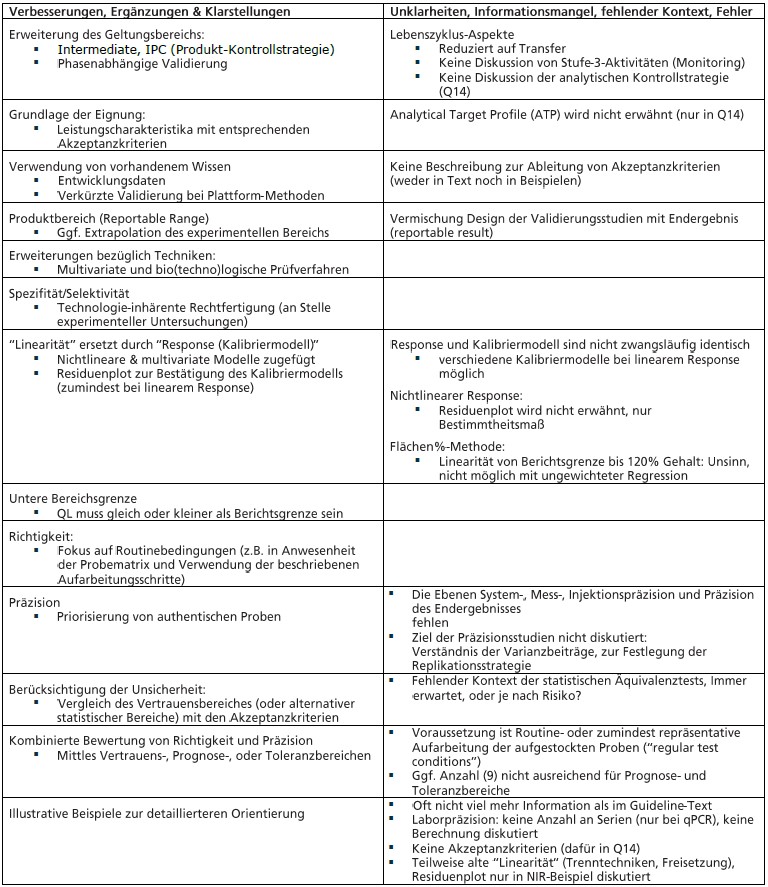
Lesen Sie im nächsten LOGFILE etwas über wichtige Aspekte bei der Umsetzung der regulatorischen Anforderungen in die Praxis.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











