

Wie lässt sich der Lebenszyklusansatz auf die Reinigungsvalidierung übertragen?
Ein gekürzter Auszug der Episode 38 aus unserem Webcast GMP & TEA
4 Min. Lesezeit | von Thomas Peither
Erschienen im LOGFILE Leitartikel 42/2023
Das Thema der 38. Episode von unserem GMP-Webcast GMP & TEA ist „Reinigungsvalidierung als Teil der Arzneimittelsicherheit“.
Im Detail gehen wir auf folgende Fragen ein:
- Wie lässt sich der Lebenszyklusansatz auf die Reinigungsvalidierung übertragen?
- Welche Anforderungen müssen Reinigungsverfahren erfüllen?
- Welche Schritte beinhaltet eine Reinigungsvalidierung?
In diesem LOGFILE konzentrieren wir uns auf die erste Frage.
Die anderen Fragen werden im GMP & TEA-Abonnement beantwortet, das im GMP & TEA-Portal abrufbar ist.
Wie lässt sich der Lebenszyklusansatz auf die Reinigungsvalidierung übertragen?
Der Lebenszyklusansatz bedeutet nichts anderes, als dass die Kontrolle über die Reinigungseffektivität fortlaufend aufrechterhalten werden muss.
Eine solche fortgesetzte Verifikation ist nicht etwa mit der Revalidierung einzelner
Bestätigungschargen abgeschlossen, sondern erfordert ein Kontrollsystem, das kontinuierlich Daten über die Wirksamkeit der Reinigung liefert.
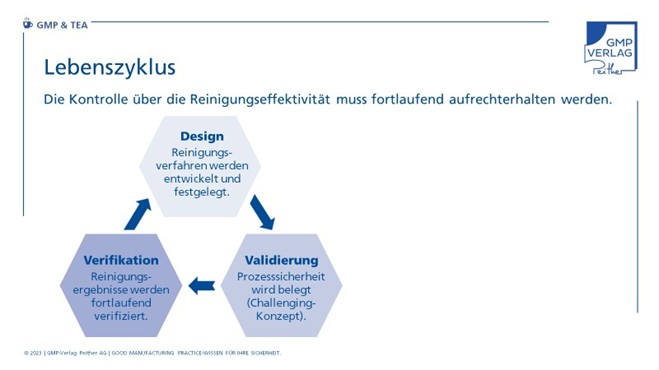
Der Lebenszyklus der Reinigungsvalidierung gliedert sich grundsätzlich in drei Phasen:
In der Designphase werden die Reinigungsverfahren für Anlagen oder Oberflächen entwickelt und festgelegt. Wird der „Quality by Design“-Ansatz verfolgt, beinhaltet dies auch die Festlegung einer Kontrollstrategie zur Steuerung und Überprüfung der Reinigungsverfahren, um reproduzierbare und spezifikationskonforme Reinigungsergebnisse zu gewährleisten.
Eine klassische chargenbezogene Reinigungsvalidierung ist dann nicht mehr erforderlich, da kontinuierlich und prospektiv sichergestellt wird, dass das Reinigungsverfahren zum gewünschten Ergebnis führt.
In allen anderen Fällen, in denen im Vorfeld keine funktionierende Kontrollstrategie etabliert wurde, muss die Eignung des Reinigungsverfahrens klassisch an einer festzulegenden Anzahl von Chargen nachgewiesen werden.
In der Validierungsphase sind dann Daten zu erheben, die die Prozesssicherheit der Reinigung belegen. Im Rahmen eines sogenannten Challenging-Konzepts lässt sich in dieser Phase zeigen, dass die gewählten Reinigungsverfahren auch unter ungünstigen, aber realen Bedingungen funktionieren.
Als letzte Phase schließt sich die eingangs erwähnte fortgesetzte Verifikation des Reinigungsergebnisses an.
Soweit die Theorie. Wie sieht es in der Praxis aus? Welche Reinigungsverfahren sind am besten geeignet? In welchem Umfang und in welchen zeitlichen Abständen sind die einzelnen Validierungsarbeiten durchzuführen?
Diese Fragen führen zum zweiten zentralen Punkt, dem Risikomanagement.

Risikoanalysen für die Auswahl der Reinigungsmittel und der zu reinigenden Anlagen sind schon seit langem gängige Praxis und sollten auch bei Ihnen der Standard sein. Seit der Revision des Annex 15 des EU-GMP-Leitfadens sind sie verbindlich vorgeschrieben.
Sie kennen den ganzheitlichen Ansatz! Je nach Prozessführung müssen neben den produktberührenden Oberflächen z. B. auch äußere Einflüsse auf das Reinigungsergebnis berücksichtigt werden.
Dieser Text ist ein Auszug aus der 38. Episode des Video-Podcasts GMP & TEA.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











