

Sechs Beispiele für Stabilitätsstudien
Ein Auszug aus dem GMP-BERATER, Kapitel 14.E.7, Studienarten
15 Min. Lesezeit | von Heike Meichsner, Dr. Olaf Mundszinger und Susanne Schweizer
Erschienen im LOGFILE 13/2021
Stabilitätsstudien sichern die gleichbleibende Qualität eines Arzneimittels und sind in allen Produktphasen ein Muss. Daher werden Stabilitätsdaten während der Entwicklungsphase, für die Zulassung und während der anschließenden Vermarktungsphase erhoben und ausgewertet.
Zentrale Aufgabe der Daten ist, Kontrollintervalle, Standzeiten und Laufzeiten festzulegen sowie Lagerungshinweise daraus abzuleiten. Im Leitartikel stellen wir Ihnen heute 6 wichtige Studientypen vor.
Langzeitstudien (long term, extended oder real time stability studies)
Bei Langzeitstudien wird der Wirkstoff oder das Arzneimittel über die gesamte geplante oder genehmigte Laufzeit unter den Langzeitbedingungen der jeweiligen Klimazone eingelagert und regelmäßig analysiert.
Man unterscheidet hier zwei Formen:
-
Langzeitstudien unter ICH-Bedingungen (Standardlagerungsbedingungen) mit vollständiger Prüffrequenz
-
jährliche Stabilitätsstudien (annual / ongoing stability study / Routine-Stabilitätsstudien) mit reduzierter Prüffrequenz
Langzeitstudien nach ICH
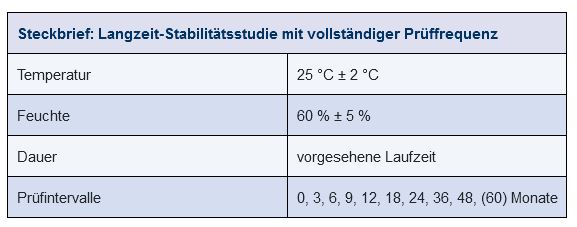
Bei Langzeitstudien unter ICH-Bedingungen wird die Prüffrequenz der ICH-Leitlinie Q1A(R2) angewendet. Die Prüfzeitpunkte liegen hier i. d. R. im ersten Jahr vierteljährlich, im zweiten halbjährlich, danach jährlich. So kommt man zu einem Prüfschema von 3, 6, 9, 12, 18, 24, 36 etc. Monaten (siehe Abbildung 14.E-22). Diese Studienart ist in folgenden Fällen erforderlich:
-
während der Entwicklungsphase an ersten Scale-up-Chargen oder Chargen, die zu klinischen Prüfungen verwendet werden
-
während der Zulassungsphase, um Stabilitätsdaten für Registrierungschargen und/oder die ersten kommerziellen Chargen zu generieren. Derartige Studien werden zur Einreichung von Zulassungsanträgen für neue Produkte / Formulierungen / Reformulierungen benötigt.
-
während der Vermarktungsphase, wenn die Zulassungsbehörde für die ersten kommerziellen Chargen eine erhöhte Datenerhebung fordert. In diesem Fall spricht man von Commitment-Studien bzw. follow up stability studies. Hierbei verpflichtet sich der Hersteller, die Ergebnisse der Studien regelmäßig an die Behörde zu berichten.
-
während der Vermarktungsphase, wenn aufgrund von Änderungen der Zulassungsunterlagen (nach Erhalt der Marktzulassung, post approval change) ein vollumfängliches Langzeitstudienprogramm erforderlich ist.
Abbildung 14.E-22 Langzeit-Stabilitätsstudie (Beispiel Klimazone I/II)
Routine-Stabilitätsstudien
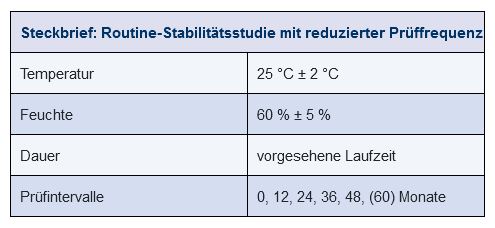
Die Stabilität eines Arzneimittels muss auch nach der Marktzulassung weiterhin regelmäßig überwacht werden. Dazu dienen die jährlichen Stabilitätsstudien mit einer reduzierten Prüffrequenz. In der Regel wird über den gesamten Lebenszyklus hinweg ein Mal pro Jahr eine Charge pro Primärpackmittel analysiert. Daraus ergeben sich jährliche Prüfzeitpunkte mit Intervallen von 12, 24, 36 Monaten etc. (siehe Abbildung 14.E-23).
Diese Vorgehensweise findet Anwendung, wenn das Produkt und sein Verhalten gut bekannt sind, so dass nur noch der Nachweis über die gleichbleibende Qualität der laufenden Produktion zu liefern ist. Man spricht in diesem Zusammenhang auch von ongoing stability (OGS) oder monitoring program.
Abbildung 14.E-23 Routine-Stabilitätsstudie (Beispiel Klimazone I/II)
Prüfungen unter intermediären Studien-Bedingungen (intermediate conditions)
Bei Prüfungen unter intermediären Bedingungen wird der Wirkstoff oder das Arzneimittel höheren Temperatur- und Feuchtewerten ausgesetzt als bei den Langzeitstudien, dafür ist die Studiendauer auf 12 Monate begrenzt. Es sollten mindestens Daten zu vier Zeitpunkten erhoben werden, d. h. es wird i. d. R. vierteljährlich analysiert. Diese Daten sind besonders wichtig, wenn Ergebnisse aus den beschleunigten Studien Qualitätsveränderungen aufweisen.
Wie die Langzeitstudien kommen diese Studien in der Entwicklungsphase, der Zulassungsphase und der Vermarktungsphase zur Anwendung.
Abbildung 14.E-24 Intermediäre Studienbedingungen (Beispiel Klimazone I/II)

Prüfungen unter beschleunigten Studien-Bedingungen (accelerated conditions)
Beschleunigte (eigentlich beschleunigende) Bedingungen liegen vor, wenn bei 40 °C/75% rF (also mindestens 10 °C über den Klimazonenbedingungen) für 6 Monate eingelagert wird und in regelmäßigen Abständen untersucht wird (z. B. 1, 3, 6 Monate). Für Zulassungs- und/oder Änderungsanträge werden gemäß der ICH-Leitlinie Q1A(R2) Ergebnisse zu mindestens drei Prüfzeitpunkten (i. d. R. 0, 3, 6 Monate) erwartet.
Durch diese Art der Prüfung wird gezeigt, welchen Einfluss kurzzeitige Temperatur- und Feuchteerhöhungen auf den Wirkstoff oder das Arzneimittel haben. Die Ergebnisse sind Basis für die Festlegung eines Lagerhinweises. Beschleunigte Bedingungen dienen somit zur Unterstützung der Langzeitdaten und gehören bei der Einreichung an die Behörde zum Datenpaket dazu.
Wie die Langzeitstudien und Studien unter intermediären Bedingungen werden diese Studien in der Entwicklungsphase, der Zulassungsphase und der Vermarktungsphase durchgeführt.
Abbildung 14.E-25 Beschleunigte Studienbedingungen (Beispiel Klimazone I/II)

Stresstests, chemische und thermische Stressbedingungen (stress testing)
Stressprüfungen werden in der Entwicklungsphase eines Wirkstoffs und in der Entwicklungsphase eines Fertigarzneimittels an der Präformulierung durchgeführt und dienen der Ermittlung von Zersetzungswegen und Identifizierung von Zersetzungsprodukten unter forcierten Bedingungen.
Chemische Stressbedingungen: Untersuchung der Zersetzungsprodukte unter alkalischen und sauren Bedingungen (Einfluss pH) oder oxidativen Bedingungen (Einfluss von Wasserstoffperoxid)
Thermische Stressbedingungen: Hier wird das Produkt, i. d. R. der Wirkstoff, bei Temperaturen von über 40 °C (in Stufen von 10 °C, also 50 °C, 60 °C etc.) und 75 % oder mehr relativer Feuchte (rF) über einen kurzen Zeitraum (wenige Tage bis einige Monate) eingelagert und geprüft.
Photostabilitätsprüfung (photostability testing)
Gemäß der ICH-Leitlinie Q1B Photostability Testing of New Drug Substances and Products gelten Photostabilitätstests als grundlegender Baustein von Stresstests. Die dem Produkt eigene photostabile Charakteristik muss damit geprüft und beschrieben werden. Eine zu erwartende Lichtbelastung darf nicht mit Einbußen bei der Qualität des Produktes verbunden sein. Dieser Test ist somit integraler Bestandteil bei der Wahl des Primärpackmittels und des Lagerhinweises.
Üblicherweise wird eine solche Studie während der Entwicklungsphase eines Wirkstoffs oder Arzneimittels einmalig an einer Charge durchgeführt. Im Falle einer gravierenden Änderung, z. B. bei der Herstellung des Wirkstoffes, bei der Formulierung des Arzneimittels oder bei der Verpackung, kann eine Wiederholung sinnvoll sein oder auch von der Behörde eingefordert werden.
Testdurchführung
Lichtquelle: Die Lichtquelle muss den Spezifikationen D65/ID65 der ISO-Norm 189091 entsprechen, wobei D65 dem internationalen Standard für Tageslicht im Freien und ID65 dem internationalen Standard für indirektes Tageslicht innerhalb eines Raums entspricht. Dafür sind Fluoreszenzröhren mit UV/Vis-Anteil, Xenon- oder Halogenlampen geeignet. Strahlungen unter 320 nm sollen mit einem geeigneten Filter eliminiert werden. Alternativ kann das Muster einer kalten, weißen Fluoreszenzlampe und zusätzlich einer Nah-UV-Lampe mit einem Spektralbereich von 320 nm bis 400 nm ausgesetzt werden.
Testbedingungen: Bei der Versuchsdurchführung ist auf die Temperaturkontrolle zu achten, damit der Lichteinfluss alleine beurteilt werden kann. Gegebenenfalls ist ein Kontrollmuster, in lichtundurchlässige Folie (z. B. Aluminiumfolie) eingepackt, mit in den Proberaum zu legen (dark control). Die Exposition muss mindestens 1200 kluxh betragen, wobei die Energie des Nah-UV-Anteils mindestens 200 Wh/m2 betragen muss.
Analyse: Nach der Bestrahlung sind die Muster auf die physikalischen und chemischen Eigenschaften zu überprüfen. Dazu gehören in erster Linie Aussehen, Gehalt und Reinheit; bei Fertigarzneimitteln werden z. B. auch Zerfallszeiten und Auflösungsgeschwindigkeit (dissolution rate) überprüft. Wenn die Prüfung Kontrollmuster beinhaltet, sollte die Analyse der belichteten Proben zusammen mit den Kontrollmustern erfolgen.
Beurteilung: Nach der Analyse sind die Veränderungen zu beurteilen. Der Entscheid richtet sich nach den Spezifikationen, die basierend auf den Erfahrungen bei der Entwicklung des Wirkstoffes oder des Fertigarzneimittels festgelegt wurden. Abschließend sind Schutzmaßnahmen (z. B. „vor Licht geschützt aufbewahren“) zu definieren, die eine Beeinträchtigung der Produktqualität verhindern helfen.
Photostabilitätsprüfung an Wirkstoffen
Bei der Versuchsdurchführung muss auf die Eigenschaften des Wirkstoffes geachtet werden, damit keine Sublimation, kein Schmelzen oder andere Veränderungen während der Bestrahlung auftreten. Der Wirkstoff (fest oder flüssig) ist in einem geeigneten Behältnis (Glas- oder Kunststoffgefäß mit lichtdurchlässiger Abdeckung) zu testen und, wo nötig, während der Durchführung zu kühlen. Es ist zu beachten, dass in der (frühen) Entwicklungsphase im Rahmen des Stresstests bewusst Photoabbauprodukte erzeugt werden, während später die Überprüfung der Schutzmaßnahmen im Vordergrund steht.
Photostabilitätsprüfung an Fertigarzneimitteln
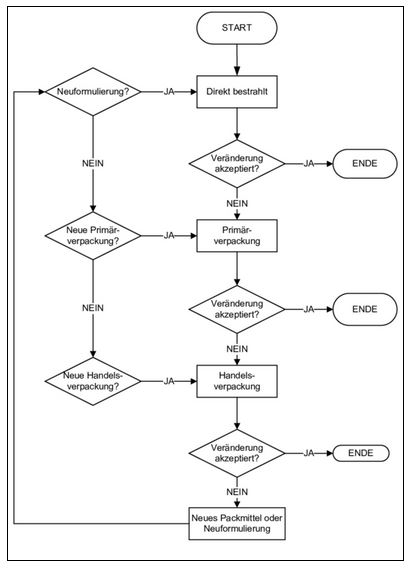
Das Fertigarzneimittel wird zuerst ohne Verpackung getestet. Wenn inakzeptable Veränderungen auftreten, wird stufenweise zuerst mit der Primärverpackung, dann auch mit der Sekundärverpackung (Handelsverpackung) geschützt. Je nach Ergebnis sind die Verpackungen zu verbessern oder auch die Formulierung zu ändern. Die Vorgehensweise ist in Abbildung 14.E-26 dargestellt. Auch hier sind wie beim Wirkstoff die Eigenschaften des Produktes zu berücksichtigen, allenfalls ist in einem geschlossenen Behältnis Fremdeinflüssen vorzubeugen.
Abbildung 14.E-26 Entscheidungsflussdiagramm für Fertigarzneimittel (Quelle: ICH Q1B)
1 ISO-Norm 10977:1993 wurde ersetzt durch 18909:2006
Schaukelstudien (freeze and thaw studies)
Schaukelstudien werden in der Regel ebenfalls in der Zulassungsphase anhand der repräsentativen Registrierungschargen durchgeführt, um das Arzneimittel kurzfristigen extremen Kältebedingungen auszusetzen. Mit ihnen kann eine unerwartete kurzfristige Belastung durch tiefe Temperaturen beurteilt werden, wie sie öfter beim Transport auftreten können.
Auch während der Entwicklung werden diese Studien häufig exemplarisch an einer Charge durchgeführt, um zu sehen, ob es unerwartete Einflüsse auf das Produkt gibt, auf die u. U. durch einen Lagerhinweis aufmerksam gemacht werden müsste. Beispielsweise neigen Kapseln nach einer gewissen Zeit zur Sprödigkeit, wenn sie tiefgefroren werden.
Eine Versuchsanordnung könnte wie folgt aussehen:
- 2 Tage bei -18 °C gefolgt von 2 Tagen bei 25 °C/60 % rF
- 2 Tage bei -18 °C gefolgt von 4 Tagen bei 40 °C/75 % rF
- 2 Tage bei 2–4 °C gefolgt von 2 Tagen bei 40 °C/75 % rF
Danach erfolgt eine Überprüfung der physikalischen Eigenschaften gegen ein unbehandeltes Muster. Hier zeigt sich in der Praxis häufig, dass tiefere Temperaturen zwar meist eine bessere chemische Stabilität bewirken, aber einen negativen Einfluss auf die physikalischen Eigenschaften (z. B. Löslichkeit) haben. Fällt während der Tiefkühlschrankphase eine Komponente (z. B. Hilfsstoff) aus, die dann nicht mehr vollständig in Lösung geht, ist die Produktqualität dauerhaft beeinträchtigt. Ein Lagerungshinweis soll Patient:innen darauf aufmerksam machen, das Produkt z. B. „nicht unter 10 °C zu lagern“.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











