
Risikoanalyse im Lebenszyklus einer Anlage
Auszug aus dem GMP:KnowHow Anlagenqualifizierung
4 Min. Lesezeit | von GMP-Verlag Peither AG
Erschienen im LOGFILE 04/2022
Vom Projektstart bis zur Freigabe des technischen Designs: Die Risikoanalyse als ständiger Begleiter.
Risikoanalysen können bzw. müssen zu verschiedenen Zeitpunkten innerhalb des Prozesses der Beschaffung und Qualifizierung durchgeführt werden. Lesen Sie einen kurzen Abriss über die Phasen der Risikoanalysen in der Projektabwicklung.
Typischerweise beginnt ein Projekt mit einer Machbarkeitsstudie, die an sich schon eine Risikoanalyse darstellt. Im nächsten Schritt (Ermittlung kritischer Prozessparameter) sollte festgehalten werden, welches Risiko von der Ausrüstung für das Produkt ausgeht. Dies sollte im Rahmen der Zusammenstellung der Anforderungen erfolgen, um die Ermittlung der kritischen Prozessparameter (Critical Process Parameters/CPP) und der kritischen Qualitätsattribute (Critical Quality Attributes/CQA) zu unterstützen.
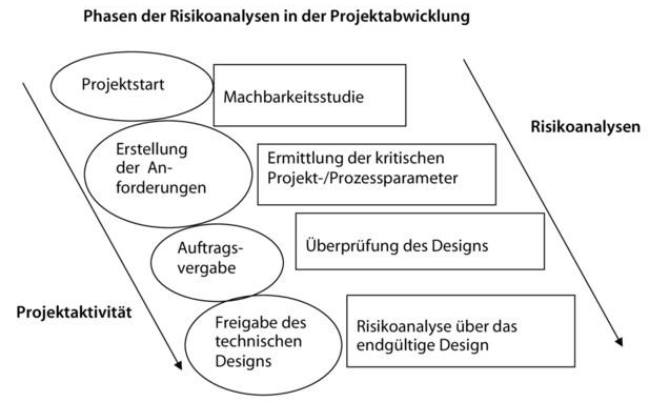
Die Einstufung der Ausrüstung in Bezug auf den Prozess kann wie folgt erfolgen:
- Critical (C): Fehlfunktion kann zur Verletzung von GMP-Anforderungen mit Einfluss auf Produktqualität und Patientensicherheit führen.
- Major (Ma): Fehlfunktion kann zu einer direkten, nicht feststellbaren Verletzung von GMP-Anforderungen ohne Einfluss auf die Patientensicherheit führen.
- Minor (Mi): Fehlfunktion kann zu einer feststellbaren Verletzung von GMP-Anforderungen ohne Einfluss auf die Patientensicherheit führen.
- Nicht anwendbar, wenn keine GMP-Anforderungen bestehen.
In der Designphase erfolgt eine Risikoanalyse, um Risiken durch Verfahrens- oder Designänderungen, Rohrleitungsführungen sowie Anordnung der Sensorik zu ermitteln und noch rechtzeitig auf das Design, die Ausrüstung und die Überwachungseinrichtungen einzuwirken. Auf Basis der Zeichnungen (R & I, Fertigungszeichnungen) und anderer Spezifikationen (Funktionsbeschreibungen, Hardware-Spezifikationen) erfolgt die Bewertung der Ausrüstung und die Festlegung der Maßnahmen zur Minimierung des Risikos.
Folgende Maßnahmen sind möglich:
- Änderung von Funktionen, Programmschritten oder Anlagenteilen
- Überprüfung in Installations-, Funktions- und Leistungsqualifizierung
- Durchführung von Studien und Versuchen im Vorfeld
- Zusätzliche Überwachungsmaßnahmen
- Regelmäßige Kalibrierung
- Instandhaltungsaktivitäten
- Administrative Maßnahmen/Adressierung in SOPs
Zur Nachverfolgung aller Maßnahmen in der Qualifizierung ist ein Vollständigkeitsnachweis (Traceability-Matrix) zu erbringen. Dies kann durch eine Referenzierung in der Risikoanalyse auf Prüfprotokollnummern, SOP-Nummern, Wartungspläne usw. erfolgen. Im Qualifizierungsbericht ist darauf zu verweisen, dass alle Maßnahmen zur Risikoreduzierung getroffen wurden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











