

Durchführung und Dokumentation der Designqualifizierung (DQ)
Dieser Text ist ein Auszug aus dem GMP-Fachwissen Qualifizierung von pharmazeutischen Produktionsanlagen.
5 Min. Lesezeit | von Thomas Peither, Ulrike Reuter, Rainer Röcker
Erschienen im LOGFILE Leitartikel 41/2020
Grundlage für die Durchführung der DQ bilden zum einen Arbeitsanweisungen und zum anderen der Validierungs-/Qualifizierungsmasterplan (siehe Kapitel 6.C.2 Validierungs-/Qualifizierungsmasterplan).
Im Rahmen einer DQ sind folgende Schritte durchzuführen:
- Erstellung eines DQ-Planes oder Beschreibung der Vorgehensweise durch eine Arbeitsanweisung oder den QMP/VMP
- Erstellung eines Lastenheftes (URS) (Kapitel 6.D.4 Benutzeranforderungsspezifikation (User Requirements Specification (URS)/Lastenheft)), ggf. erste Risikoanalyse (Kapitel 6.D.3 Risikoanalysen in der Design-Phase)
- Vergleich des Pflichtenheftes (Kapitel 6.D.7 Pflichtenheft) mit dem genehmigten Lastenheft (URS), z. B. in Form einer Traceability Matrix (Kapitel 6.D.9 Tracematrix (Vollständigkeitsnachweis)) oder mit Hilfe einer Checkliste
- Lieferantenqualifizierung
- Vergleich der vorgelegten Konstruktionsunterlagen und der Funktionsbeschreibung (Spezifikationen) des technischen Systems inkl. Steuerung mit den Anforderungen des Lasten- und Pflichtenheftes
- Durchführung, Dokumentation und Genehmigung der DQ
Die Durchführung der DQ begleitet die Planung und Konstruktion des technischen Systems (vgl. Abbildung 6.D-2 ).
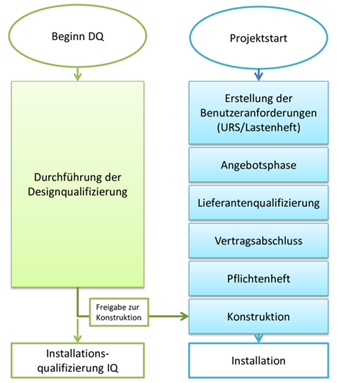
Abbildung 6.D-2 Designqualifizierung im Projektablauf
Mitunter ist in der Praxis zu beobachten, dass sich die Aktivitäten ausschließlich auf den Vergleich von Lasten- und Pflichtenheft beschränken. Diese Überprüfung deckt aber nur einen kleinen Teil der DQ-Aktivitäten ab und kann so nicht den gewünschten Effekt erbringen, z. B. das Erkennen von Konstruktions- und Entwicklungsfehlern in einer frühen Projektphase. Somit besteht weiterhin die Möglichkeit, dass Fehler bis zur IQ und OQ unerkannt bleiben und deren Beseitigung dann deutlich umfangreichere Ressourcen (Zeit, Personal) erfordert. Deshalb ist es wichtig, kritische Punkte schon im Vorfeld zu erkennen. Auch alle weiteren Spezifikationen und technischen Dokumente, die während der Designphase und später im Lebenszyklus einer Anlage auftreten, sind auf die Einhaltung der Anforderungen aus dem Lastenheft (URS) zu überprüfen.
Lieferantenqualifizierung
Gerade bei Anlagen mit komplexen Steuerungs-, Antriebs- und Visualisierungssystemen ist eine Auditierung und Qualifizierung der potentiellen Lieferanten unumgänglich. Bevor die Entscheidung für einen Lieferanten getroffen wird, muss überprüft werden, ob dieser definierte Prozesse zur Sicherstellung der Qualität im Unternehmen installiert hat. Ein ISO-9001-Zertifikat alleine reicht nicht aus. Vielmehr muss gewährleistet sein, dass die GMP-Anforderungen an einen Pharmalieferanten erfüllt werden.
Gerade bei Anlagen mit hohem Softwareanteil sind oftmals auch Sublieferanten im Einsatz, die sehr genau zu betrachten sind. Was passiert, wenn solche Firmen in ein paar Jahren nicht mehr am Markt sind? Haben diese bei der Softwareerstellung denselben Qualitätsstandard wie renommierte Firmen im GMP-Umfeld?
Ob man Audits vor Ort durchführt oder postalisch, hängt von der Komplexität und Kritikalität des Systems ab. Dazu sollte man z. B. überlegen, welche Risiken für den Patienten, die Produktsicherheit und die Datenintegrität bestehen.
Dabei sollten ein Auditplan und ein dazugehöriger Fragenkatalog als roter Faden dienen.
Die nachfolgenden Aspekte sollten unbedingt hinterfragt werden:
- Hat der Lieferant ein funktionierendes QMS installiert?
- Wie wird dessen ordnungsgemäße Durchführung sichergestellt?
- Wie wird der Umgang mit Unterlieferanten geregelt, vor allem bei der Softwareentwicklung?
- Gibt es eine dokumentierte Vorgehensweise und Standards speziell in der Softwareerstellung?
- Gibt es einen Lebenszyklus für die Softwareentwicklung bis hin zur Stilllegung der Anlage?
- Ist das Abnahmeprozedere ausreichend beschrieben?
- Gibt es Referenzkunden?
- Wie viele Anlagen hat das Unternehmen schon im pharmazeutischen Umfeld installiert?
Weiterführende Informationen zu dieser Thematik finden Sie in Kapitel 18.H Lieferantenqualifizierung.
Erforderliche Dokumente
Wie in Abbildung 6.D-3 gezeigt, sind während der DQ folgende Dokumente zu erstellen (vgl. auch Kapitel 6.C Qualifizierungsdokumentation):
- genehmigter DQ-Plan (falls Vorgehensweise nicht schon detailliert in einer Arbeitsanweisung festgelegt ist)
- Risikoanalyse zur GMP-Einstufung des technischen Systems und des damit verbundenen Prozesses; Festlegung der kritischen Qualitätsattribute bzw. kritischen Prozessparameter
- Benutzeranforderungen/User Requirements Specification (URS)/Lastenheft (oder vergleichbares Dokument)
- Pflichtenheft/Spezifikationen (oder vergleichbares Dokument)
- Risikoanalyse zur Einstufung der GMP-kritischen Funktionen, Systemelemente und Messstellen
- Konstruktionszeichnungen
- Funktionsbeschreibungen für Anlage und Steuerung
- Risikoanalyse (z. B. als FMEA) zur Überprüfung des Entwurfs (Design), der Funktionen/Prozessschritte zur Entdeckung von Fehlern im Prozess
- Kategorisierung der einzelnen Softwaresysteme nach GAMP 5©
- DQ-Bericht mit Bewertung von Abweichungen
- gegebenenfalls weitere Dokumente
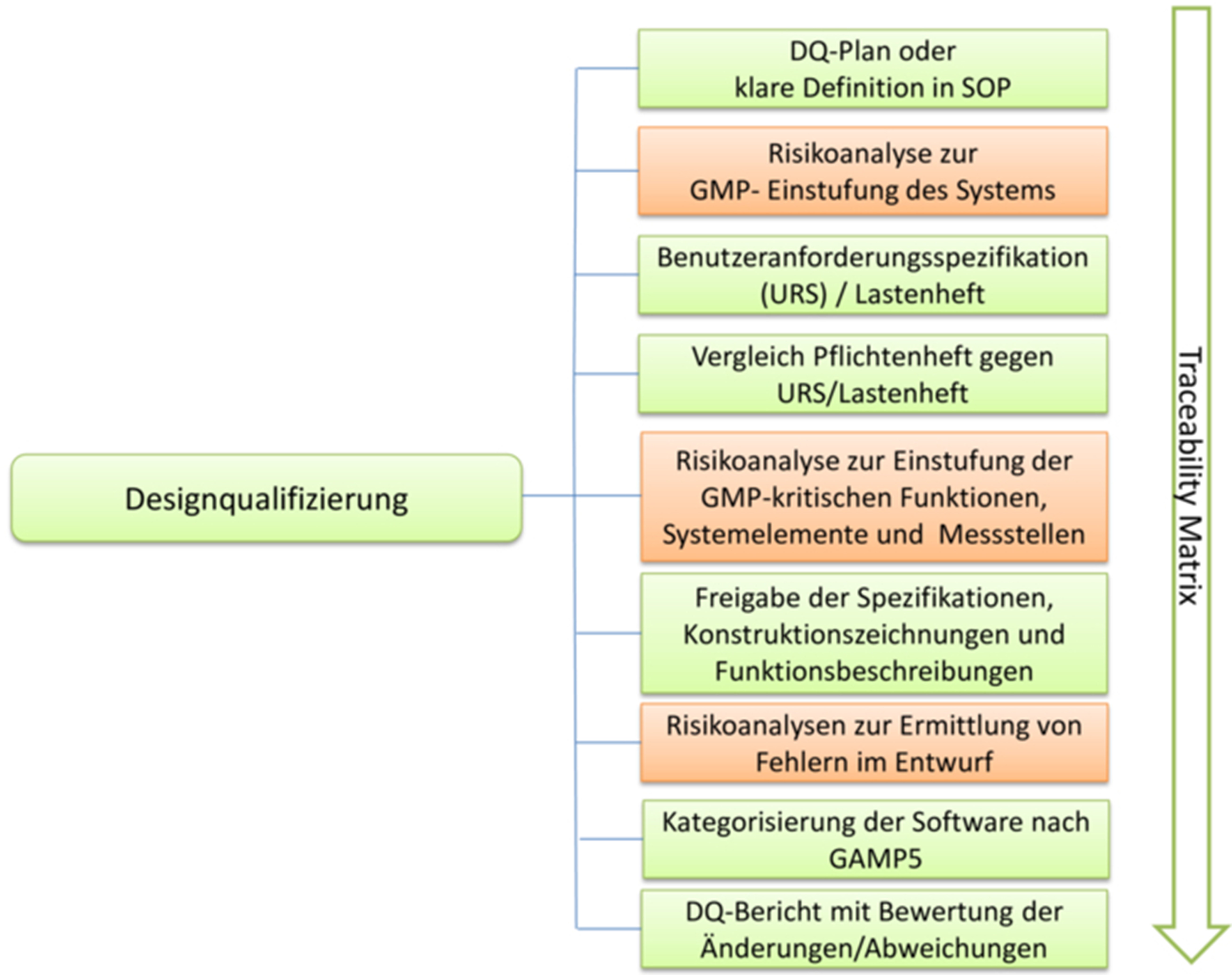
Abbildung 6.D-3 Durchführung und Dokumentation der Designqualifizierung
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











