

Führen von Logbüchern
Ein Auszug aus der SOP-403 Protokollführung, Logbücher und Laborjournale
5 Min. Lesezeit | von Dr. Christine Oechslein
Erschienen im LOGFILE 38/2020
Logbücher dienen dazu, eine lückenlose Dokumentation über Einsatz und Zustand von Räumen, Maschinen, Geräten oder Beständen (z. B. Standards, Reagenzien, Chemikalien) zu schaffen.
Darum sind vom Betriebspersonal stets die nachfolgend aufgelisteten Daten bzw. Vorkommnisse in zeitlicher Reihenfolge in Raum- und Anlagen-Logbücher einzutragen. Für Reagenzien- und Standard-Logbücher sind die nachfolgend beschriebenen Regeln analog anzuwenden, soweit sinnvoll.
- Belegung (Produktbezeichnung, auch technische Versuche, Qualifizierungsläufe oder nicht-GMP-Nutzung)
- (Funktions-)Störungen, Defekte
- außergewöhnliche Beobachtungen
- Reinigung
- Kalibrierung
- Justierung
- Prüfung
- Wartung, Serviceleistungen aller Art
- Reparatur, ersetzte Teile
- Änderungen am Gerät (geplante und ungeplante)
- Änderung des GxP-Status
- (Wieder-)Inbetriebnahme („Freigabe zur GMP-konformen Nutzung“)
- Außerbetriebnahme
- Verkauf oder Verschrottung
Einzutragen sind jeweils
- Art, Datum und Dauer des Ereignisses (mit Uhrzeit Beginn/Ende)
- Querverweis auf die Nummer des zugehörigen Protokolls (z.B. Chargendokumentation, Reinigungsprotokoll, Wartungsbericht, Reparaturbeleg, Change-Control-Nummer)
- ggf. Bemerkungen oder Beobachtungen
- Datum und Namenszeichen der ausführenden Person
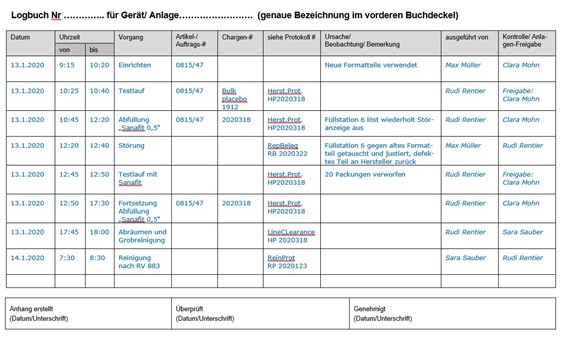
Ein Beispiel für ein Anlagen- und Geräte-Logbuch findet sich in Anlage 3 zu dieser SOP. Je nach Geräte-Typ (z. B. Analysengerät, Monitoringinstrument, Versorgungsanlage) sind die Spalten für die Pflicht-Eintragungen anzupassen. Verantwortlich für die sachdienliche Anpassung ist der Raum- bzw. Geräte-Verantwortliche zusammen mit dem Leiter der betreffenden Abteilung.
Auch Arbeiten, die auf elektronischem Weg erfasst werden, wie beispielsweise Wartungen, müssen im Logbuch eingetragen werden. Ausführliche Bemerkungen oder Ergebnisse sind in diesem Falle jedoch nicht erforderlich. Als Querverweis muss die vom elektronischen (Wartungs-)System vergebene Positions- oder Auftrags-Nummer im Logbuch eingetragen werden.
Für das Führen von „klassischen“ gebundenen Logbüchern gibt es einige Alternativen. In vielen Firmen werden Belegung (Produktfolge) und Reinigung separat in elektronischen Dokumentationssystemen erfasst. Auch technische Aufzeichnungen zu Wartung, Reparaturen, Kalibrierungen usw. werden oftmals in separaten Datenbanken gesammelt. Es ist nicht erforderlich, diese Informationen zusätzlich komplett (d. h. redundant) in einem elektronischen oder papierbasierten Logbuch zu führen. Entscheidend ist, dass die jeweiligen Aufzeichnungssysteme validiert sind und Umgang und Zugriffsberechtigungen detailliert in SOPs beschrieben und geschult sind.
Zusätzlich muss eine Möglichkeit bestehen, die in den verschiedenen Systemen erfassten Ereignisse im Bedarfsfall (z. B. bei Fehlerursachenanalysen) in eine chronologische Reihenfolge zu bringen. Oftmals ist eine einzeilige Eintragung im Logbuch mit einem Querverweis auf die Referenznummer der elektronischen Dokumentation dazu die einfachste Möglichkeit.
Bei der virtuellen Firma Maas und Peither Pharma AG wird ein konventionelles Logbuch verwendet. Eine weitere Möglichkeit ist das Führen von Statuskarten, die im aktuellen Zeitraum direkt an der Anlage geführt werden und dann in das Logbuch überführt (eingeklebt) werden. Damit wird oftmals die Compliance seitens der Mitarbeiter erhöht.
Bei komplexen Anlagen können pro Einzelkomponente separate Logbücher (elektronisch oder Papier) bzw. Statuskarten geführt werden. Wichtig ist jedoch auch hier, dass im Bedarfsfall tatsächlich alle Ereignisse, die die Gesamtanlage betreffen, schnell und problemlos im Gesamtüberblick betrachtet werden können.
Sofern für das Führen von Logbüchern oder Statuskarten umfangreichere Erklärungen erforderlich sind, die den Rahmen dieser Protokollführungs-SOP sprengen, z. B. zu den verschiedenen Anlagenzuständen („frei für GMP-konforme Nutzung“, „in Wartung“ usw.), dann sollten diese Vorgaben separat in einer eigenen „Logbuch-SOP“ beschrieben werden.
Für alle diese Eintragungen gelten die unter Punkt 6.2– 6.6 genannten Protokollführungsregeln.
Das Betriebspersonal ist dafür verantwortlich, vor jeder Nutzung eines Raums oder Geräts anhand des Logbuchs zu prüfen, ob er/es in sauberem und betriebsbereitem Zustand ist. Anschließend wird der Beginn der Nutzung in das Logbuch eingetragen und das Logbuch wieder an seinem anlagennahen Aufbewahrungsort hinterlegt. Bei Abschluss der jeweiligen Arbeiten ergänzt das Betriebspersonal die Angaben im Logbuch mit allen o. g. Daten und Beobachtungen. Gerätebezogene Protokolle, Wartungsbelege, Reparaturbelege oder ähnliche Unterlagen legt der ausführende Mitarbeiter abschließend in der Anlagendokumentation ab (Standort: siehe vorderer Buchdeckel des Logbuchs).
Der jeweilige Raum- bzw. Geräteverantwortliche ist „Owner“ des Logbuchs. Er legt das erste Logbuch im Rahmen der IQ-Phase an (Vergabe der Logbücher durch den Archiv-Verantwortlichen der QS) und überprüft danach regelmäßig Vollständigkeit und Richtigkeit der Eintragungen im Logbuch sowie die zugehörigen in der Anlagendokumentation hinterlegten Rohdaten. Er überprüft insbesondere, dass auch von externen Personen (Wartungs-, Kalibrier-, Qualifizierungs- oder andere Service-Dienstleister) durchgeführte Arbeiten so protokolliert werden, dass die Standards dieser SOP erfüllt werden.
Jedes Logbuch ist nummeriert und paginiert. Geräte-/Raumlogbücher enthalten als fixe Informationen folgende Angaben:
- Innenseite des vorderen Buchdeckels:
- Logbuch-Nummer
- Inventarnummer des Geräts/des Raums
- Falls das Gerät Teil eines komplexen technischen Systems ist: Bezeichnung des Systems, zu dem das Gerät gehört.
- Geräteverantwortlicher und Stellvertreter
- gegebenenfalls Standort des Geräts (Gebäude/Raum)
- zuständige technische Servicefunktion
- gegebenenfalls zuständiger Qualifizierungsbeauftragter
- Standort der Anlagendokumentation gemäß SOP-600_A7
- Innenseite des hinteren Buchdeckels:
- Liste aller Personen, die im Logbuch eintragen: voller Name, Namenskürzel
Zur Vergabe von Logbüchern und zu ihrer Archivierung siehe Punkt 6.10.3 und 7.
Anlage 3: Beispiel für ein Logbuch
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de










