

Häufiger GMP-Mangel: unzureichende Statuskennzeichnung von Reinigungsmaterial
Auszug aus dem GMP-BERATER, Kapitel 21.E.2.8, Unzureichende Kennzeichnung zu Status/Verwendbarkeit von Reinigungsmaterial
7 Min. Lesezeit | von Lea Joos
Erschienen im LOGFILE 29/2022
Der Mangelpunkt: Im Reinigungsraum waren für verschiedene Reinigungs- und Desinfektionsmittel keine Aufbrauchsfristen nach Anbruch angegeben.
Außerdem konnte gereinigtes Reinigungsequipment nicht von ungereinigtem Reinigungsequipment unterschieden werden. Z. B. befanden sich zwei Kisten mit Wischtüchern im Reinigungsraum. In welcher Kiste sich die gereinigten und in welcher die ungereinigten Wischtücher befanden, war nicht erkennbar.
(Ref.: EU-GMP-Leitfaden Teil I Nr. 3.37)
Das war das Problem
Reinigungs- und Desinfektionsverfahren müssen wirksam sein. Dazu werden geeignete Materialien ausgewählt und die Verfahren validiert. Die Reinigung und Desinfektion dürfen selbst keine Kontaminationsquelle darstellen.
Im vorliegenden Fall wurden Reinigungs- und Desinfektionsmittel auch nach Anbruch weiterverwendet. Das Anbruchsdatum war nicht angegeben. Die Haltbarkeiten nach Anbruch waren der Firma für diese Reinigungs- und Desinfektionsmittel nur teilweise bekannt. Insofern war nicht durchgehend nachvollziehbar, ob eventuell vorliegende Haltbarkeiten nach Anbruch eingehalten wurden.
Werden Reinigungs- oder Desinfektionsmittel nach Anbruch weiterverwendet oder über die vom Hersteller angegebene Haltbarkeit hinaus verwendet, obwohl eine Weiterverwendung beispielsweise die Gefahr einer Verkeimung birgt, kann dies statt zur Reinigung zur Kontamination der Flächen oder des Personals beitragen.
Ähnlich verhält es sich mit der Verwendung von Reinigungsmaterialien. Werden verunreinigte Reinigungsmaterialien wie Wischtücher, Bürsten oder Schwämme in der Reinigung eingesetzt, stellen diese ebenfalls eine Kontaminationsquelle dar. Deswegen muss hier sichergestellt werden, dass keine Verwechslungsgefahr zwischen gereinigten und verunreinigten Reinigungsmaterialien besteht, sondern diese eindeutig gekennzeichnet bzw. als gereinigt oder verunreinigt erkennbar sind.
Das ist der häufigste Fallstrick
Für jedes Reinigungs- und Desinfektionsmittel unterschiedliche Aufbrauchsfristen festzulegen, wird von den Firmen häufig als Herausforderung und teilweise, z. B. in der Herstellung von nicht-sterilen Arzneimitteln, auch als „nicht notwendig“ wahrgenommen. Gerade im nicht-sterilen Bereich wird häufig der Aufwand als viel größer wahrgenommen, als der mögliche Nutzen.
Die Kennzeichnung von gereinigtem und verunreinigtem Equipment wird für Produktionsausrüstung oft durchgeführt, aber nicht für Reinigungsausrüstung. Das liegt daran, dass es einen zusätzlichen Dokumentations- und Organisationsaufwand bedeutet, den überwiegend externes, häufig wechselndes Reinigungspersonal leisten und die Firma (aufgrund des zeitlichen Aufwands) zusätzlich bezahlen müsste – ohne dass die Firma den direkten Nutzen dieser Maßnahme erkennt.
So vermeiden Sie diesen Fehler
Die Reinigung unterliegt insbesondere bei manueller Durchführung einer großen Bandbreite an Einflussgrößen. Um die Variabilität zu verringern, werden diese Einflussgrößen in Verfahrensanweisungen festgelegt und in der Reinigungsvalidierung auf Wirksamkeit überprüft.
Dabei werden häufig Variablen als „gegeben“ angesehen, die nicht ausreichend festgelegt sind.
Wenn Sie z. B. ein wiederaufbereitetes Wischtuch oder eine Reinigungsbürste in der Validierung verwenden müssen, dann sind die einzelnen wiederaufbereiteten Wischtücher oder Reinigungsbürsten vielleicht untereinander z. B. hinsichtlich der Reinigungskraft nicht vergleichbar.
Vor der Verwendung wiederaufbereiteter Wischtücher müssen die mit der Wiederaufbereitung verbundenen Risiken in einer Risikoanalyse identifiziert und bewertet werden. Mögliche Risiken sind beispielhaft in Abbildung 21.E-20 dargestellt:
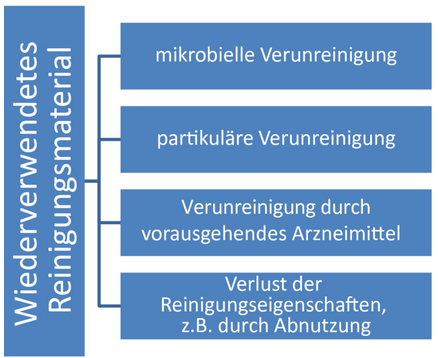
Abbildung 21.E-20 Risiken der Wiederverwendung bereits verwendeter und verunreinigter Reinigungsmaterialien
Ein Teil dieser Risiken kann durch eine Validierung z. B. des Wiederaufbereitungsverfahrens reduziert werden, z. B. eine mikrobielle Verunreinigung. Der Verlust der Reinigungseigenschaften z. B. durch Abnutzung ist jedoch ein Risikofaktor, der nicht durch ein validiertes Wiederaufbereitungsverfahren minimiert werden kann. Er muss daher im Rahmen der Reinigungsvalidierung berücksichtigt werden: reinigt mein Reinigungsmaterial nach der ersten, sechsten oder x-ten Wiederaufbereitung/Verwendung immer gleich gut?
Bei der anschließenden Überprüfung der „Reinigungskraft“ des wiederaufbereiteten oder wiederverwendeten Reinigungsmaterials stellen Sie in der Reinigungsvalidierung vielleicht fest, dass Sie dieses zehnmal aufbereiten oder verwenden können, bevor die „Reinigungskraft“ zu stark nachlässt und den Reinigungserfolg beeinträchtigt. Diese validierte Bedingung muss anschließend überwacht werden.
Vielleicht stellen Sie auch fest, dass für den überprüften Reinigungsschritt eine Überwachung der Wiederaufbereitungszyklen nicht erforderlich ist.
Dennoch werden Sie die Reinigung im Rahmen der Reinigungsvalidierung vermutlich mit sauberen Reinigungsmaterialien durchführen und nicht mit verunreinigten. Und damit müssen Sie auch im „Reinigungsalltag“ sicherstellen, dass die validierte Bedingung „zur Reinigung wird gereinigtes Reinigungsmaterial verwendet“ eingehalten werden kann.
Lassen Sie bei der Überwachung der Einhaltung der validierten Bedingungen Ihrer Fantasie freien Lauf:
-
Wir brauchen eine Separierung von gereinigtem und verunreinigtem Reinigungsmaterial – wie ist das am pragmatischsten möglich?
-
Wir müssen die Anzahl der Reinigungszyklen des Wischtuchs überwachen – welche Möglichkeiten gibt es und welche ist praktisch machbar?
Reden Sie dazu auch mit dem betroffenen Personal: dem Reinigungspersonal, der Wiederaufbereitungsfirma, den Personen in der Firma, die damit unmittelbar zu tun haben.
Dann findet sich sicherlich eine kreative Lösung, die den zeitlichen Aufwand in Grenzen hält, z. B. der Einsatz von Klebepunkten zur Überwachung der Wiederaufbereitungszyklen oder verschiedenfarbige Boxen für reine und unreine Wischtücher.
Ähnliche Fragen sollten Sie sich für Reinigungs- und Desinfektionsmittel stellen, die nach Anbruch weiterverwendet werden sollen: Wenn der Hersteller keine Angaben macht oder ich das Reinigungs- und Desinfektionsmittel auch nach der vom Hersteller angegebenen Aufbrauchsfrist verwenden möchte: reinigt und desinfiziert das Reinigungs- und Desinfektionsmittel nach Anbruch, nach 6 Wochen oder nach 12 Monaten immer gleich gut?
Und wenn Sie im Rahmen der Reinigungsvalidierung eine wirksame Desinfektion bis zu vier Monate nach Anbruch festgestellt haben: überwachen Sie anschließend die Einhaltung dieser validierten Bedingung – anhand der für Sie praktikabelsten Lösung.


Abbildung 21.E-21 Nachvollziehbarkeit der Statuskennzeichnung von Reinigungsmaterial
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











