

Qualitätsrisikomanagement (QRM) und GDP
Auszug aus dem GMP:KnowHow Pharmalogistik (GDP), Kapitel 1.5, Qualitätsrisikomanagement
8 Min. Lesezeit | von Simone Ferrante
Erschienen im LOGFILE 19/2022
Der Gesetzgeber verlangt, dass jedes Unternehmen selbst herausfindet, welche und wie viele Qualitätssicherungsmaßnahmen für seine Produkte (auch Dienstleistung) und seine Mitarbeiter, Anlagen und Räume erforderlich sind.
So eine systematische Untersuchung, Bewertung, Lenkung, Kommunikation und Überprüfung des Qualitätsrisikos (inklusive bestehender Chancen) nennt man Qualitätsrisikomanagement (QRM).

Das QRM folgt dem Grundgedanken einer vorbeugenden Fehlerverhütung anstelle einer nachsorgenden Fehlererkennung und -korrektur durch frühzeitiges Erkennen und Bewerten etwaiger Fehlerursachen. Anfallende Kontroll- und Fehlerfolgekosten in der Produktentwicklung und Prozessplanung werden vermieden und die Kosten insgesamt gesenkt. Das Hauptziel von QRM nach GMP und GDP ist, die Patientensicherheit nicht zu gefährden. Daher geht die Anwendung von QRM über die Produktentwicklung und Prozessplanung hinaus, denn die Produktqualität eines Arzneimittels oder Medizinprodukts muss während des gesamten Lebenszyklus gewährleistet sein.
Das QRM dient nicht nur zur Ermittlung von Risiken, sondern auch von Chancen. Verbesserungspotentiale sind ebenfalls risikobasierend aufzudecken. (ISO 9001:2015)
Das Risikomanagement sollte eine zentrale Stellung im Qualitätsmanagement haben.
Patientensicherheit
Das QRM soll schon frühzeitig dafür sorgen, dass nahezu alle kritischen Punkte im Kontext mit der Validierung, der Festlegung von Spezifikationen und Nutzen-Risiko-Abwägung eines Arzneimittels (oder von Produkten allgemein) diskutiert und entschieden werden. Die Bewertung der Qualitätsrisiken muss auf wissenschaftlichen Erkenntnissen beruhen und immer vor dem Hintergrund der Patientensicherheit gesehen werden.
Verfahrenssicherheit und Prozessverständnis
Die Qualität des Produktes kann nicht am Ende hineingeprüft werden, sondern muss im Laufe der gesamten Handhabe sichergestellt sein. Aber auch nach der Herstellung des Produktes können sich verschiedene Einflussfaktoren negativ auf das Produkt auswirken. Daher ist die Sicherstellung der Qualität auch erforderlich bei Transport und Lagerung und als fortlaufender Prozess anzusehen.
Es ist wichtig, die kritischen Punkte und Schritte des gesamten Prozesses zu kennen. Durch die Aufdeckung der kritischen Punkte wird Transparenz geschaffen. Wissenschaftlich basierte Schlussfolgerungen werden kommuniziert. Dies steigert das Verständnis der Mitarbeiter und erlaubt eine gezielte Optimierung der Prozesse.
Durch die gezielte Durchführung einer Risikoanalyse können bei der Zusammenfassung der hieraus abgeleiteten, notwendigen Maßnahmen die vorhandenen Ressourcen zielgerichtet und sinnvoll eingesetzt werden. Die Priorisierung von Risiken hilft dabei. Zur Erfassung und Bewertung der Risiken sollten Standards geschaffen werden. Dadurch können ähnliche Prozesse besser verglichen und Synergien genutzt werden.
Beispiele der Prozesse und Anwendungen eines Qualitätsrisikomanagements findet man unter anderem in den EU-Leitlinien zur Guten Herstellungspraxis (GMP Teil III). Hier wurde die Leitlinie Q9 der Internationalen Harmonisierungskonferenz („ICH“) aufgenommen.
Wie wird QRM in der Praxis eingesetzt?
Es gibt sehr verschiedene Möglichkeiten und Methoden, Qualitätsrisikomanagement in der Praxis einzusetzen. Hier einige Beispiele:
- bei der Planung von Qualifizierung und Validierung, um prospektiv zu ermitteln und zu begründen, welche Anlagenteile bzw. Parameter als kritisch zu betrachten sind
- bei der Erstellung von Trainingsplänen, um die Häufigkeit und Lerninhalte für bestimmte Schulungsgruppen festzulegen
- bei der Erstellung von Hygieneplänen, um Methode und Frequenz von Reinigung und ggf. Desinfektion festzulegen
- bei der Planung von Transportwegen, um prospektiv die für die Arzneimittelqualität kritischen Parameter und notwendigen Maßnahmen festzulegen
- im Rahmen von Änderungskontrolle (Change Control), um die geplanten Änderungen in Risikoklassen einzuteilen und die notwendigen Folgemaßnahmen festzulegen
- bei der Bearbeitung von Abweichungen, Reklamationen, Beanstandungen (die das Produkt selbst betreffen) und Retouren, um retrospektiv mögliche Gefahren für die hergestellten Produkte zu erkennen und prospektiv geeignete Korrekturmaßnahmen festzulegen.
Ein Risiko ist die Kombination aus der Eintrittswahrscheinlichkeit eines Schadens und dem Ausmaß dieses Schadens. Prozesse bzw. Prozessschritte werden systematisch hinsichtlich ihrer Kritikalität bewertet und daraus angemessene Maßnahmen zur Risikokontrolle und Risikominimierung entwickelt.
Welche besonderen Gefahren drohen in der Supply Chain?
Auf dem Vertriebsweg von pharmazeutischen und medizinischen Produkten kann viel passieren, z. B:
- Beschädigung oder Kontamination der Ware beim Umgang und während des Transportes
- Nicht-Einhaltung von speziellen Transport- und/oder Lagerungsbedingungen, wie Temperatur und, wenn gefordert, Luftfeuchte
- falsche Kennzeichnung von Ware
- Verlust von Ware
- Verwechslung von Ware
- Einbringen von gefälschten Arzneimitteln in die legale Lieferkette
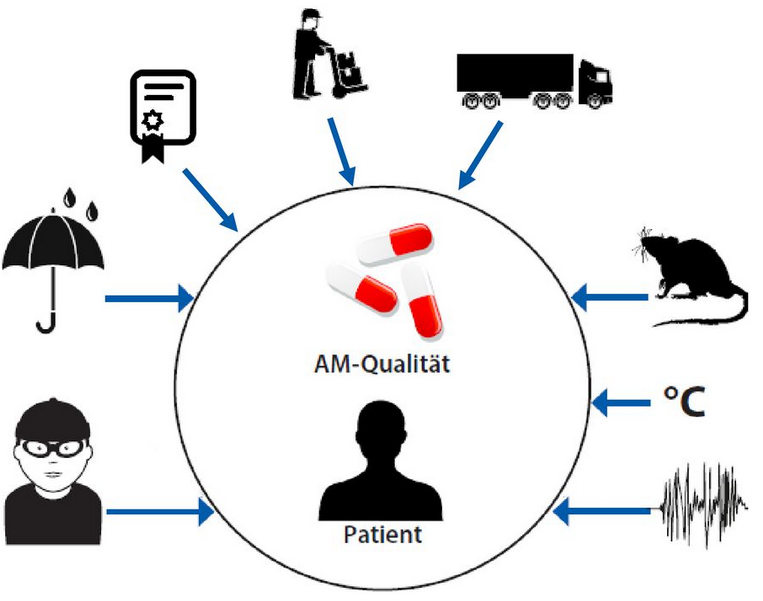
Abbildung: GDP hat den Schutz vor äußeren Einflüssen zum Ziel (Quelle: GDP-Kompaktwissen (modifizierte Darstellung))
Kontaminationen
Kontaminationen sind Verunreinigungen, die von überall her in das Produkt gelangen können. Sie müssen unbedingt vermieden werden, da sie die Qualität des Arzneimittels mindern und zu vorzeitigem Verderb führen können. Wahrscheinliche Gefahrenquellen im Distributionsbereich sind Staub und Feuchtigkeit.
Verunreinigungen mit anderen Wirkstoffen oder Produkten treten dagegen eher selten auf – eigentlich nur, wenn bei den Routineabläufen Arzneimittelpackungen beschädigt werden.
Auch Schädlinge können zur Verunreinigung und Verderb von Produkten führen. Ein umfassendes Schädlingsbekämpfungsprogramm (Pest Control) ist zur Risikominimierung gefordert.
Weil Verunreinigungen immer einen Weg finden und es so viele unterschiedliche Quellen dafür gibt, sind sehr viele Schutzmaßnahmen notwendig, die alle gleichzeitig befolgt werden müssen.

Verwechslungen
Fertigarzneimittel sehen auf den ersten Blick oft gleich aus. Eine Verwechslung von zwei Produkten oder von zwei Dosierungen desselben Wirkstoffes beim Versand kann für den Patienten lebensbedrohliche Folgen haben, sofern Apotheker oder Arzt sie nicht rechtzeitig entdecken. Eine chargenreine Lagerung, eindeutige Beschriftungen, elektronische Bestandsführung und Verifikations-Scans sollen das Risiko einer Verwechslung ausschließen.
Menschen mit korrupten Absichten – Diebe/Fälscher
Leider gibt es auch im pharmazeutischen und medizinischen Umfeld Menschen mit korrupten Absichten. Dies war 2011 Anlass für die Veröffentlichung der EU-Richtlinie 2011/62/EU zum Fälschungsschutz von Arzneimitteln. Als Folge wurden u. a. die GDP-Regeln weltweit verschärft. Seitdem müssen Vorkehrungen für die Aufrechterhaltung der Legalität der Lieferkette unternommen werden. Sämtliche Lieferanten und Zulieferer sind zu qualifizieren. Zutritts- und Zugriffsschutz auf Dokumentation und Ware muss während des gesamten Lebenszyklus der Produkte sichergestellt sein.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











