

Regulatorische und normative Vorgaben zur Abwehr von Arzneimittelfälschungen
Auszug aus dem GMP-BERATER, Kapitel 21.I, Abwehr von Arzneimittelfälschungen
8 Min. Lesezeit | von Dr. Stephan Schwarze
Erschienen im LOGFILE Leitartikel 02/2020
Die Richtlinie 2011/62/EU des Europäischen Parlaments und des Rates vom 8. Juni 2011 zur Änderung der Richtlinie 2001/83/EG (sogenannte EU Falsified Medicines Directive) wurde innerhalb der EU durch nationale Gesetzgebung in nationales Recht transponiert.
In dieser Richtlinie sind zwei Sicherheitsmerkmale definiert:
- eine Kodierung mit einem individuellen Erkennungsmerkmal (unique identifier) in Form eines zwei-dimensionalen Kodes, der eine eindeutige Produktidentifikationsnummer mit einer randomisierten Seriennummer kombiniert (2D-Data-Matrix-Code)
- das Anbringen einer Vorrichtung gegen Manipulation (anti-tampering device), wobei hier keine weiteren Details zur Art der Vorrichtung festgelegt werden.
Die konkreten Festlegungen bezüglich der Ausgestaltung der Sicherheitsmerkmale, insbesondere der Kodierung, finden sich in der Delegierten Verordnung (EU) 2016/161der Kommission vom 2. Oktober 2015 zur Ergänzung der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates durch die Festlegung genauer Bestimmungen über die Sicherheitsmerkmale auf der Verpackung von Humanarzneimitteln (siehe Kapitel G.1.2). Diese Delegierte Verordnung ist direkt geltendes Recht in der EU. Aufgrund der Komplexität des Themas wurde zusätzlich ein Frage-Antwort-Dokument bereitgestellt mit dem Titel „Safety Features for Medicinal Product for Human Use – Questions and Answers“ (siehe Kapitel G.1.2.1). Die Veröffentlichung zahlreicher Versionen innerhalb kürzester Zeit und der dabei immer weiter zunehmende Inhalt (siehe Abbildung 21.I-8) zeigen, wie komplex die Materie ist. Einerseits, weil es regelmäßig eine Diskrepanz zwischen einem von Juristen formulierten Gesetzestext und dessen praktischer Interpretation und Umsetzung gibt. Andererseits wurden offensichtlich nicht alle relevanten Aspekte eindeutig genug geregelt, so dass diese Lücken nachträglich geschlossen werden müssen.

Abbildung 21.I-8 „Safety Features for Medicinal Product for Human Use – Questions and Answers“ – Entwicklung der Anzahl der Kapitel, Seiten sowie der Fragen und Antworten
Die Auswahl einer geeigneten Vorrichtung gegen Manipulation obliegt allein dem Hersteller. Ziel ist, dass diese Originalitätsverschlüsse eine Manipulation des Inhalts einer Arzneimittelverpackung erschweren und einen Eingriff in die Integrität der Arzneimittelverpackung visuell erkennbar machen. Detailliertere Festlegungen zu deren Auswahl finden sich nicht in den regulatorisch relevanten Dokumenten oder Richtlinien. In diesem Zusammenhang wird im Frage-Antwort-Dokument auf die einschlägige Norm verwiesen, derzeit noch auf die CEN EN 16679:2014 [1] (weitere Details dazu siehe unten).
Am 30. November 2018 wurde jedoch von der International Organization for Standardization (ISO) die ISO-Norm 21976:2018 mit dem Titel „Packaging – Tamper verification features for medicinal product packaging“ [2] veröffentlicht, die auf der o. a. EN-Norm basiert. Es ist zu erwarten, dass diese praktisch inhaltsgleiche ISO-Norm in naher Zukunft die Europanorm ersetzen wird. Eine systematische Übersicht der Zusammenhänge zwischen Sicherheitsmerkmalen, deren Ausprägungen und Zielgruppen ist in Abbildung 21.I-9 dargestellt.
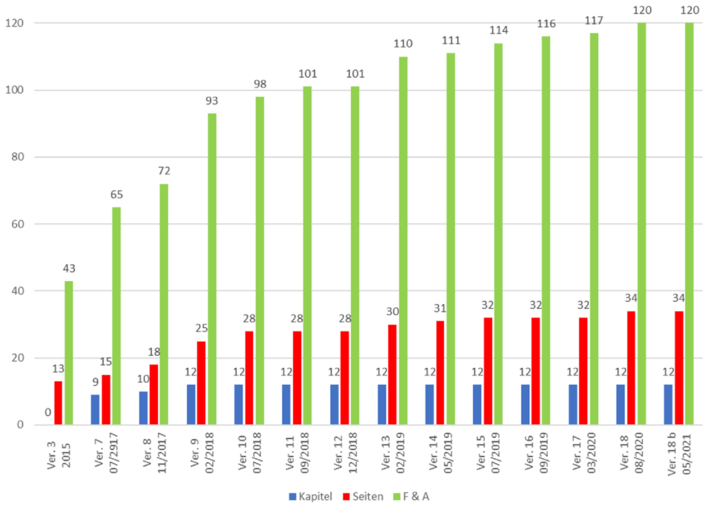
Abbildung 21.I-9 Systematik der Sicherheitsmerkmale und Zielgruppen
Fälschungssichere Merkmale können ebenso wie individuelle Kodierungen prinzipiell auf Sekundär- und Primärbehältnissen, bei bestimmten Arzneiformen auch auf dem Arzneimittel selbst eingesetzt werden. Originalitätsverschlüsse hingegen sind nur für Sekundär- und Primärbehältnisse üblich.
Eine schematische Darstellung für die praktische Umsetzung auf einer Sekundärverpackung (hier auf einer Faltschachtel) zeigt Abbildung 21.I-10.
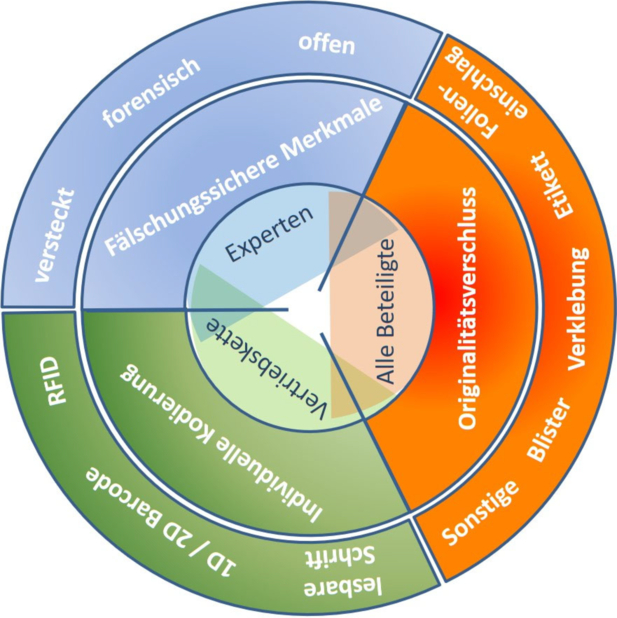
Abbildung 21.I-10 Schematische Darstellung von Sicherheitsmerkmalen auf einer Faltschachtel
[1] DIN EN 16679:2015-03, Verpackung – Merkmale zur Überprüfung von Manipulationen an Arzneimittelverpackungen; Deutsche Fassung EN 16679:2014
[2] ISO 21976:2018, Packaging -- Tamper verification features for medicinal product packaging
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











