

Rückstellmuster – GMP-konform bereitstellen und aufbewahren
Ein Auszug aus der SOP-602-02, Kapitel 6.1 und 6.2
7 Min. Lesezeit | von Dr. Stephanie Blum
Erschienen im Logfile 45/2022
Gemäß Anhang 19 des EU-GMP-Leitfadens müssen Rückstellmuster von allen Ausgangsstoffen, Primärpackmitteln und bedruckten Packmitteln sowie von Fertigarzneimitteln und klinischen Prüfpräparaten aufbewahrt werden.
Wie Sie GMP-konform die Rückstellmusteraufbewahrung organisieren und Rückstellmuster bereitstellen, zeigt Ihnen ein Auszug aus der Muster-SOP 602-02 Bereitstellung und Aufbewahrung von Rückstellmustern.
Arbeitsablauf und Verantwortlichkeiten – Grundsätze
Rückstellmuster werden von allen Ausgangsstoffen (ausgenommen Lösungsmittel, Gase und Wasser), Primärpackmitteln und bedruckten Packmitteln sowie von Fertigarzneimitteln und klinischen Prüfpräparaten aufbewahrt.
Die Analyse oder Überprüfung von Rückstellmustern erfolgt z. B. im Zusammenhang mit Reklamationen, etwaigen Rückrufen und Fragen der Arzneimittelsicherheit. Sie kann wertvolle Informationen zur Sicherheit, Wirksamkeit und Qualität des betreffenden Arzneimittels liefern.
Die nachfolgende Tabelle gibt einen Überblick über die unterschiedlichen Aufbewahrungsfristen von Rückstellmustern:
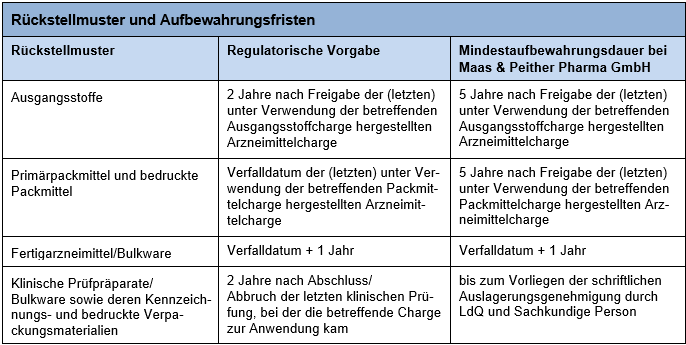
Insbesondere im Hinblick auf Ausgangsstoffe und Packmittel gibt es zahlreiche Möglichkeiten, die Rückstellmusteraufbewahrung zu organisieren.
Natürlich kann die Aufbewahrung aller Rückstellmuster einfach entsprechend den regulatorischen Vorgaben erfolgen. Die bei Maas & Peither Pharma GmbH für Rückstellmuster von Ausgangsstoffen und Packmitteln definierten Vorgaben (siehe Tabelle oben) gehen teilweise über diese regulatorischen Anforderungen hinaus. Der Vorteil liegt darin, dass so die Rückstellmuster-Aufbewahrungsdauer vereinheitlicht und dadurch deren Überwachung vereinfacht wird.
Durch die einheitliche Vorgabe "5 Jahre nach Freigabe" für die Rückstellmuster-Aufbewahrung von Ausgangsstoffen und Packmitteln ist sichergestellt, dass die regulatorisch für Rückstellmuster von Packmitteln vorgegebene Frist sicher eingehalten wird. (Die Haltbarkeit von Arzneimitteln ist auf maximal 5 Jahre begrenzt.) Übrigens: Rückstellmuster bedruckter Packmittel müssen nicht zwangsläufig separat gezogen und aufbewahrt werden, sondern können auch einfach Bestandteil der Ansichts- bzw. Referenzmuster des betreffenden Fertigarzneimittels sein.
Alternativ zum hier vorgestellten Prozedere oder der Aufbewahrung gemäß regulatorischen Vorgaben kann aber auch eine konkrete Aufbewahrungsfrist für Rückstellmuster von Ausgangsstoffen und Packmitteln definiert werden. Dies bietet sich insbesondere dann an, wenn die Umschlagszeiten dieser Materialien vergleichsweise kurz und relativ einheitlich (oder per SOP standardisiert) sind. Eine solche Aufbewahrungsfrist könnte sich z. B. aus der maximalen Material-Umschlagszeit plus der regulatorisch vorgegebenen Rückstellmuster-Aufbewahrungsfrist errechnen oder (darauf basierend) ganz allgemein auf x Jahre nach Wareneingang festgelegt werden.
Noch ein Hinweis: Rückstellmuster von Ausgangsstoffen dürfen kürzer als hier angegeben aufbewahrt werden, vorausgesetzt in den Zulassungsunterlagen ist eine kürzere Haltbarkeit der betreffenden Ausgangsstoffe angegeben.
Bereitstellung von Rückstellmustern
Die Bereitstellung repräsentativer Rückstellmuster einschließlich der erforderlichen Mengen ist in material- bzw. produktspezifischen Probenahmeplänen geregelt. Probenahme, Verpackung und Kennzeichnung der Rückstellmuster erfolgen entsprechend diesen Probenahmeplänen sowie gemäß den Vorgaben der jeweils geltenden Probenahme-SOPs (SOP-203: Probenahme von Ausgangsstoffen und Packmitteln, SOP-204: Probenahme von Zwischenprodukten, Bulkware und Fertigprodukten).
Die Rückstellmuster werden von folgenden Mitarbeitern bereitgestellt und zur Einlagerung an die Qualitätskontrolle (Gruppe: Stabilitätsprüfungen/Rückstellmuster) übergeben:
- Ausgangsstoffe und Packmittel: Mitarbeiter der Qualitätskontrolle (Gruppe: Probenahme)
- Fertigarzneimittel: autorisierte Mitarbeiter der Produktion (Gruppe: Verpackung)
- Klinische Prüfpräparate (einschließlich Kennzeichnungs- und bedruckter Verpackungsmaterialien): autorisierte Mitarbeiter aus F&E (Gruppe: Bereitstellung klinischer Prüfpräparate)
Die Probenahme ist eine Aufgabe der Qualitätskontrolle. Ablauftechnisch ist es aber oft sinnvoll und daher üblich, dass die Probenahme direkt von den an der Herstellung beteiligten Mitarbeitern vorgenommen wird. Bei Maas & Peither Pharma GmbH wurden daher mehrere Mitarbeiter der entsprechenden Abteilungen zur Probenahme autorisiert und dementsprechend geschult. Diese Delegation der Probenahme wurde schriftlich vom LdQ genehmigt und ist außerdem Bestandteil der Stellenbeschreibung der jeweiligen Mitarbeiter.
Referenzmuster
Referenzmuster werden in einer Menge gezogen, die ausreichend ist für zwei komplette Vollanalysen gemäß genehmigter Prüfvorschrift. Die hierfür jeweils konkret erforderliche Menge wird im Rahmen der Methodenvalidierung/-etablierung von der Qualitätskontrolle ermittelt und im entsprechenden Probenahmeplan festgehalten.
Referenzmuster werden von jeder Charge der folgenden Materialien und Produkte gezogen:
- Ausgangsstoffe
- Primärpackmittel
- bedruckte Packmittel
- Fertigarzneimittel oder deren zugehörige primärverpackte Bulkware
- klinische Prüfpräparate oder deren zugehörige primärverpackte Bulkware
Ansichtsmuster
Von jeder Fertigarzneimittelcharge wird eine vollständig verpackte Einheit einschließlich Packungsbeilage als Ansichtsmuster gezogen.
Von Prüfpräparaten werden keine Ansichtsmuster des fertig verpackten Produkts gezogen/aufbewahrt; die erforderliche Information zur Sekundärverpackung und Etikettierung sowie Muster der Packmittel, Etiketten und sonstiger Kennzeichnungsmaterialien können der jeweiligen Verpackungsdokumentation entnommen werden.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











