

Temperatur- und Umgebungskontrolle
Auszug aus dem GMP:KnowHow Pharmalogistik (GDP)
8 Min. Lesezeit | von Simone Ferrante
Erschienen im LOGFILE 06/2022
„Da die Lagertemperatur und die Luftfeuchte entscheidenden Einfluss auf die Haltbarkeit der eingelagerten Materialien haben, muss ein Lagerraum vor seiner Verwendung qualifiziert werden“, betont Pharmalogistikexpertin Simone Ferrante im GMP:KnowHow Pharmalogistik (GDP).
Ein Auszug aus dem neuen Wissensportal gibt wichtige Hinweise zum unverzichtbaren Temperaturmapping und -monitoring. Sie erfahren auch, was Sie bei Erreichen des Warn- oder gar des Aktionslimits tun müssen.
Basierend auf den Spezifikationen eines Arzneimittels bzw. Medizinprodukts ist genau festgelegt, welche äußeren Bedingungen für das jeweilige Produkt eingehalten werden müssen.
Eine Nicht-Einhaltung dieser Umgebungsbedingungen kann gravierende Auswirkungen auf das Produkt haben. Um die Patientensicherheit zu gewährleisten, muss jedoch ein definierter, angestrebter Zustand des Produktes aufrechterhalten werden. Dieser wird durch Qualifizierung und Validierung nachgewiesen. Vor allem äußere Einflüsse können dazu führen, dass festgelegte Werte nicht eingehalten werden können.
Der gesamte Produktlebenszyklus eines Arzneimittels steht unter einer lückenlosen Überwachung. Nur so kann gewährleistet werden, dass der Endverbraucher ein Produkt mit der geforderten Qualität erhält.

Abbildung 1: Inhalte einer Umgebungskontrolle
Die Umgebungskontrolle beinhaltet Kontrollmaßnahmen für risikobehaftete Zustände. Für die pharmazeutische Industrie gelten sehr strenge Vorschriften für die Durchführung von Kontrollen, da gewährleistet sein muss, dass das Produkt keinen negativen Einflüssen ausgesetzt ist. Diese „strengen“ Anforderungen gelten weiterhin für die gesamte Supply Chain und somit ebenfalls für den Logistikdienstleister. Die Umgebungskontrolle soll insbesondere sicherstellen, dass:
- Räume, Anlagen und Geräte sauber und in einem hygienischen Zustand sind,
- geforderte Temperatur- und Luftfeuchtigkeitsbereiche eingehalten werden,
- keine unautorisierten Personen sich in den Räumlichkeiten aufhalten,
- und alle Bereiche frei von Ungeziefer sind.
Temperaturverteilstudien/Temperaturmapping
Da die Lagertemperatur und die Luftfeuchte entscheidenden Einfluss auf die Haltbarkeit der eingelagerten Materialien haben, muss ein Lagerraum vor seiner Verwendung qualifiziert werden.
Jedoch ist der Parameter „Luftfeuchtigkeit“ bei der Lagerung von Fertigarzneimitteln aufgrund der geprüften Verpackung eher von untergeordneter Bedeutung.
An verschiedenen Orten im Lager werden Thermometer, Hygrometer oder Datalogger platziert, die bei verschiedenen Witterungsbedingungen (Sommer/Winter) zeitgleich über mehrere Tage die Lagerbedingungen aufzeichnen (Temperaturmapping). Temperaturmappings sind ein bedeutender Bestandteil der Lagerqualifizierung.
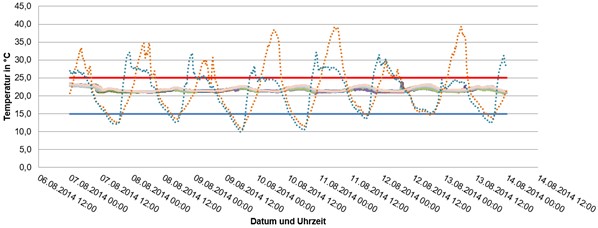
Abbildung 2: 7 Tage Temperaturmapping Sommer im Hochregallagerbereich
Die Anzahl und die Standorte der Messgeräte sind risikobasiert auf die Größe und Einrichtung des Lagerbereiches abzustimmen und die Akzeptanzkriterien (oberer und unterer Grenzwert) zu definieren. Naturgemäß ist die Anzahl der für ein Temperaturmapping eingesetzten Messpunkte deutlich größer als beim Routinemonitoring. Solche Messungen sind nicht nur im leeren Lager durchzuführen, sondern auch bei laufendem Betrieb und gefülltem Lager (unter repräsentativen Bedingungen). Gerade in klimatisierten Lagerräumen kann die Temperaturverteilung bei voll beladenen Regalen ganz anders sein als im leeren Zustand. Ständig offene Rolltore führen im Winter beispielsweise unerwartet zu Frostschäden an Arzneimitteln.
Bestenfalls kann in einem Mapping gezeigt werden, dass diese Grenzwerte nicht über- bzw. unterschritten werden. Bei Abweichungen muss deren Ursache ermittelt werden und Maßnahmen müssen getroffen werden, um diese zu minimieren. Ist dies nicht möglich, so darf an der Stelle kein Arzneimittel gelagert werden oder die Lagerung ist besonders streng zu überwachen.
Die Ergebnisse der Temperaturverteilung entscheiden darüber, welche Positionen der Logger für das Routinemonitoring der Lagerbedingungen genutzt werden. Sinnvollerweise sind dies die Messpunkte mit den größten Temperaturschwankungen.
Je nach Ergebnis können sich diese Messstellen im Sommer von denen im Winter unterscheiden. Beispielsweise kann die Sonneneinstrahlung im Sommer den Logger an der festgelegten Position so beeinflussen, dass keine realistischen Messungen möglich sind. Weisen alle Messpunkte unkritische Schwankungen in der gleichen Größenordnung auf, so ist eine gleichmäßige Verteilung der Datenlogger für das Routinemonitoring zulässig.
Monitoring der Lagertemperatur und Luftfeuchte
Auch nach erfolgreicher Lagerqualifizierung müssen die Lagerungsbedingungen (Temperatur, wo erforderlich auch Luftfeuchte) regelmäßig überprüft und protokolliert werden.

Abbildung 3: Temperatur- und Luftfeuchtigkeitslogger der Fa. Testo SE & Co. KGaA
Für das Monitoring genutzt werden können z. B.
- manuell auszulesende Datenlogger, die die Werte in festgelegten Intervallen messen, oder
- Datenlogger, die ihre Daten automatisch an einen Rechner übermitteln, und/oder
- die zur Erfassung der Messwerte, Alarme und ggf. eingeleiteter Maßnahmen mit der zentralen Gebäudeleittechnik verbunden sind.
Weichen die Messungen von den vorgeschriebenen Temperatur- oder Feuchtebereichen ab, so muss die Qualitätssicherung informiert werden, damit die Abweichung bewertet und Korrekturmaßnahmen ergriffen werden können.
Auf Basis der Qualifizierungs-Ergebnisse wird das Routinemonitoring ausgerichtet. Hierfür wird häufig ein elektronisches Monitoringsystem eingesetzt. Dieses System muss validiert sein. Die unteren Abbildungen zeigen Messungen von einem solchen Monitoringsystem. Falls vordefinierte Grenzwerte überschritten werden, werden Alarme ausgelöst. Üblicherweise wird zwischen Warnlimit und Aktionslimit unterschieden. Beispielsweise kann festgelegt werden, dass der Grenzwert gleich dem Aktionslimit ist und das Warnlimit X Grad vor dem Aktionslimit liegt. Die Variable „X“ wäre risikobasierend zu definieren.
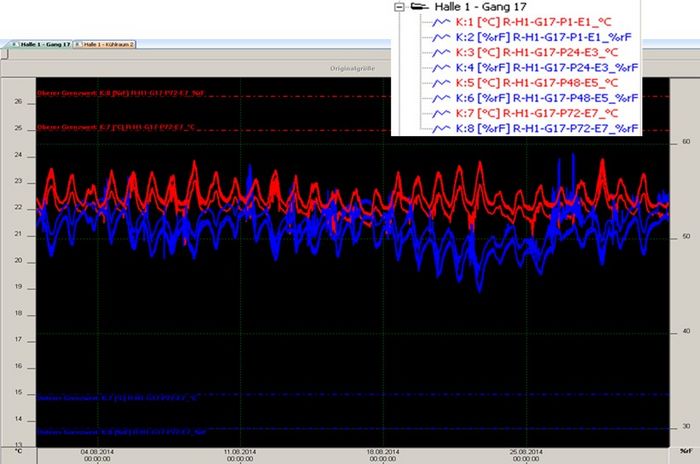
Abbildung 4: Temperaturmonitoring bei Raumtemperatur
Abbildung 4 zeigt Messwerte von Temperatur- und Luftfeuchtigkeit von Datenloggern, die in unterschiedlichen Höhen eines Hochregals im Lagerbereich 15–25°C angebracht wurden. Die linke Ordinate gibt die Temperaturskala, die rechte Ordinate die Feuchteskala an. Messwerte zur Temperatur sind in rot und Messwerte für Feuchte sind in blau dargestellt. Grenzwerte sind mit 15 und 25°C sowie 30 und 65 %rel. Feuchte hinterlegt.

Abbildung 5: Temperaturmonitoring in einem Kühllager
Abbildung 5 zeigt Temperaturmessungen von verschieden Datenloggern (unterschiedliche Farbe) eines Kühllagers. Der festgelegte Soll-Bereich liegt zwischen 2°C und 8°C.
Bei Änderungen an dem Monitoringsystem ist der valide Zustand zu berücksichtigen. Veränderungen an den Räumlichkeiten oder der Temperaturüberwachungsanlage sind über einen Änderungsantrag zu beantragen. Eine Revalidierung kann nach der Änderung verlangt werden.
Maßnahmen bei Erreichen des Warnlimits
Bei Überschreiten des Warnlimits wird eine Meldung an verantwortliche Personen gesendet. Die Art und Weise der Meldung ist zu definieren. Die Meldungsempfänger sind bei Erhalt verpflichtet, auf direktem Wege das System zu überprüfen und falls erforderlich zu intervenieren. Es muss gewährleistet werden, dass die Temperatur sich nicht weiter von den vorgegebenen Grenzen entfernt.
Maßnahmen bei Erreichen des Aktionslimits
Bei Überschreiten des Aktionslimits wird ebenfalls eine Meldung an verantwortliche Personen gesendet. Diese sind bei Erhalt verpflichtet, auf direktem Wege Maßnahmen einzuleiten, um die Temperatur schnellstmöglich in den geforderten Bereich zurück zu bringen.
Ebenso ist die Verantwortliche Person gemäß GDP in Kenntnis zu setzen und die Abweichung über das Abweichungsmanagement zu bearbeiten.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











