

Unzureichende Reinigungsvalidierung
Gekürzter Auszug aus dem GMP-BERATER, Kapitel 21.E.2.5, Unzureichende Reinigungsvalidierung
5 Min. Lesezeit | von Lea Joos
Erschienen im LOGFILE Leitartikel 45/2020
Der Mangelpunkt
Bei der Reinigungsvalidierung wurden zur Umsetzung der EMA-Guideline die PDE-Werte der Wirkstoffe ermittelt.
Dabei wurde bei einem Kombinationsarzneimittel nur der höher dosierte Wirkstoff betrachtet, da in der Risikobewertung davon ausgegangen wurde, dass der niedriger dosierte Wirkstoff bei Abreinigung des höher dosierten Wirkstoffs auf ein akzeptables Maß nicht mehr nachweisbar ist. In diese informelle Risikobetrachtung war weder der PDE noch die Reinigbarkeit oder Löslichkeit des niedriger dosierten Wirkstoffs eingeflossen. Die bisherige Worst-Case-Substanz wurde ohne weitere Rationale beibehalten.
Außerdem waren aus den PDE-Gutachten folgende Punkte nicht nachvollziehbar:
- Umfang bzw. Vollständigkeit der Literaturrecherche mit Angabe der verwendeten Daten
- Auswahlkriterien des für die Berechnung verwendeten LOEL
Ref.: EU-GMP-Leitfaden Teil I Nr. 5.21 und Annex 15 Nr. 10.6)
Das war das Problem
Der Mangel gestaltete sich zweiteilig:
Erstens: Unzureichende Einbindung der PDE-Werte in das Worst-Case-Konzept der Reinigungsvalidierung
Der PDE war im vorliegenden Fall nicht für alle Wirkstoffe berechnet worden, weil die Firma davon ausging, dass nach der Abreinigung von Wirkstoff A bis zu dessen Grenzwert von Wirkstoff B „nichts mehr vorhanden“ ist, so dass eine PDE-Berechnung für Wirkstoff B von der Firma als nicht notwendig erachtet wurde.
Die Annahme, ein Wirkstoff sei „nicht mehr vorhanden“ ist grundsätzlich schwierig: wie viele Nachkommastellen in welcher Einheit bedeuten, dass „nichts“ mehr vorhanden ist?
Eine Beispielrechnung soll dieses Problem darstellen (siehe Abbildung 21.E-11):
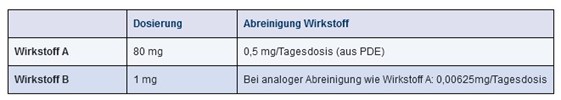
Abbildung 21.E-11 Annahme einer vergleichbaren Abreinigung von Wirkstoff A und Wirkstoff B
Heißt 0,00625 mg (oder 6,25 µg), dass „nichts“ mehr vorhanden ist?
Darauf würden Sie hoffentlich antworten: das hängt davon ab. Und zwar von der Potenz/Toxizität des Wirkstoffs B, also vom PDE-Wert. Und damit sind wir beim eigentlichen Problem: die Bewertung, ob ein „noch so niedriger“ Wert nach der Reinigung niedrig genug ist, kann nur mittels des PDE erfolgen – und der fehlte in diesem Fall.
Außerdem war von der Firma auch nicht weiter hinterfragt worden, ob denn Wirkstoff B tatsächlich „gleich gut“ abgereinigt wird und damit dieser Analogieschluss überhaupt möglich ist. So könnte Wirkstoff A z. B. sehr gut wasserlöslich sein, Wirkstoff B hingegen sehr schlecht – damit würden die beiden Substanzen bei der Reinigung ein unterschiedliches Verhalten aufweisen.
Unabhängig von den verschiedenen weiteren PDE-Werten für die Wirkstoffe C und D war der bislang als Worst-Case-Wirkstoff definierte Wirkstoff A weiterhin als Worst-Case-Wirkstoff definiert. Eine Bewertung der weiteren Gültigkeit der Einstufung als Worst-Case-Wirkstoff auf Basis der berechneten PDE-Werte für die Wirkstoffe A, C und D fehlte.
Zweitens: Mängel im PDE-Gutachten
Aus dem PDE-Gutachten selbst war nicht erkennbar, welche Datenbanken konkret zur Literaturrecherche verwendet worden waren. Es konnte daher nicht beurteilt werden, ob die Literaturrecherche vollständig im Sinne der EMA-Guideline erfolgte (siehe Kapitel H.3.3.7 und Kapitel 8.E.2.1 Der PDE und seine Ableitung – Grundlagen einer Risikobewertung).
Das PDE-Gutachten ermittelte den PDE-Wert für Wirkstoff A aus dem LOEL (Lowest Observed Effect Level). Die EMA-Guideline fordert jedoch zunächst die Ermittlung des NO(A)EL (siehe Kapitel H.3.3.7). Wird nicht der NO(A)EL verwendet oder existieren mehrere NO(A)ELs, muss begründet werden, warum welcher Wert für die weitere Berechnung herangezogen wird. Diese Begründung fehlte.
So vermeiden Sie diesen Fehler
Zum ersten Teil des Mangels:
Für die Einbindung der PDE-Werte in die Reinigungsvalidierung sollte ein konkretes Konzept existieren, das folgende Fragen beantworten können sollte:
- Für welche Stoffe müssen PDE-Gutachten erstellt werden? Und für welche vielleicht nicht? Welche Kriterien werden dafür angelegt?
- Können verschiedene Wirkstoffe für das PDE-Gutachten in einer Gruppe zusammengefasst werden? Was wäre hierfür erforderlich?
- Wie fließt der PDE-Wert in die Herleitung des Worst-Case-(Wirk-)Stoffs ein? Welche Kriterien fließen neben dem PDE mit ein? In welcher Gewichtung? Gibt es Kriterien, die „immer“ zu einer Betrachtung im Rahmen der Validierung führen müssen?
- Können PDEs/ADEs aus toxikologischen Betrachtungen in einem Nicht-Arzneimittel-Zusammenhang übertragen werden auf Arzneimittel? Welche (Mindest-)Kriterien müssen erfüllt sein?
Ein Ansatz zur Ermittlung des Worst-Case-Produkts ist in Kapitel 8.D Festlegung des Validierungsumfangs dargestellt. Hier sind als Auswahlkriterien beispielsweise Wirkstoffgehalt, Löslichkeit und therapeutische Dosis aufgeführt. Weitere Kriterien, die bislang ebenfalls Verwendung fanden, sind beispielsweise Reinigbarkeit und Toxizität. Gerade der Punkt „Toxizität“ kann nun sehr gut mit dem Ergebnis des PDE-Gutachtens untermauert werden und sollte entsprechend in die Auswahl des Worst-Case-Produktes einfließen. Dabei muss die Gewichtung der verschiedenen Auswahlkriterien risikobasiert erfolgen, wie in Abbildung 21.E-12 beispielhaft dargestellt wird.
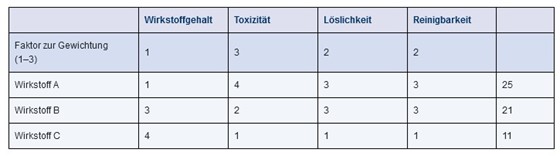
Abbildung 21.E-12 Beispiel einer Gewichtung von Auswahlkriterien für die Worst-Case-Substanz
In diesem Beispiel wird eine Bewertung von 1 bis 4 vorgenommen mit folgender Bedeutung:
- Wirkstoffgehalt: sehr niedrig (1) bis sehr hoch (4)
- Toxizität: sehr niedrig (1) bis sehr hoch (4)
- Löslichkeit: sehr gut (1) bis sehr schlecht (4)
- Reinigbarkeit: sehr gut (1) bis sehr schlecht (4)
Für die Auswahlkriterien erfolgt eine Gewichtung mit den Faktoren 1 bis 3. Die Gewichtung der Toxizität mit dem Faktor 3 bedeutet, dass der jeweilige Toxizitätswert mit 3 multipliziert wird, um die Toxizität stärker in die Endauswertung einfließen zu lassen.
Beispiel:
- Ohne die Gewichtung beträgt die Summe der Bewertungsfaktoren für die Wirkstoffe A und B jeweils 11;
- unter Berücksichtigung der Gewichtungsfaktoren erhält man für Wirkstoff A 25 und für Wirkstoff B 21; die Toxizität fließt also stärker in die Endauswertung ein.
Für eine solche Bewertung ist eine Gruppierung der PDE-Werte notwendig, z. B. PDE 1–50 µg/Tag zu „Toxizität hoch“ (Gruppe 3). Solche Gruppierungen können im Einzelfall die Kritikalität eines Wirkstoffes nicht ausreichend wiedergeben. In solchen Fällen muss der Wirkstoff aufgrund eines Einzelkriteriums, z. B. des PDE-Werts oder der Löslichkeit allein als Worst-Case-Wirkstoff betrachtet werden.
Für einzelne Wirkstoffe keinen PDE-Wert berechnen zu lassen, ist sehr schwer begründbar. Denn in diese Begründung muss neben anderen Kriterien wie Wirkstoffgehalt und Reinigbarkeit eben auch die Toxizität einfließen. Und wie untermauern Sie die – wenn nicht über den PDE-Wert?
Dazu vielleicht ein kleiner Ausflug in den US-amerikanischen Raum. Im 21 CFR 211 finden Sie an mehreren Stellen das Satzelement scientifically justified (wortwörtlich: wissenschaftlich begründet/gerechtfertigt). Behalten Sie dieses Schlagwort im Kopf, wenn Sie beispielsweise Wirkstoffe von der PDE-Berechnung ausschließen.
 Egal was Sie tun – fragen Sie sich vorher:
Egal was Sie tun – fragen Sie sich vorher:
Habe ich dafür wirklich eine Begründung, die auf wissenschaftlichen Daten basiert?
Für Aktivitäten, die Sie entsprechend SOPs oder anderen Vorschriften, wie beispielsweise Herstellvorschriften, durchführen, ist diese wissenschaftliche Begründung in der Regel gegeben. Aber sobald Sie sich in einem darüber hinaus gehenden Bereich befinden, denken Sie an diesen Merksatz.
Zum zweiten Teil des Mangels:
Die Firma sollte eigene Anforderungen an externe toxikologische Gutachten formulieren, die von „Nicht-Toxikologen“ überprüft werden können. Dabei geht es im Prinzip um eine „logische“ Nachvollziehbarkeit der Angaben des Toxikologen insbesondere zu folgenden Bereichen:
- Art und Umfang der Literaturrecherche mit Angabe der Datenbanken
- Kriterien zur Ableitung des relevantesten NO(A)EL oder Begründung der Verwendung eines LO(A)EL
- Kriterien zur Ableitung der Korrekturfaktoren für die PDE-Berechnung
Dann können auch nicht-toxikologische Mitarbeiter die Einhaltung dieser Kriterien prüfen und grobe Fehler z. B. hinsichtlich der Literaturrecherche oder der nachvollziehbaren Dokumentation ausschließen.
Haben Sie Fragen oder Anregungen? Bitte schreiben Sie uns: redaktion@gmp-verlag.de











