

APS-Invalidierung und First Air im Fokus
Bericht über die PDA Good Aseptic Manufacturing Conference 2026
4 Min. Lesezeit | von Dr. Sabine Paris
Erschienen im LOGFILE 13/2026
Auf der PDA Good Aseptic Manufacturing Conference 2026 in Frankfurt erörterte Alberto Gonzales (Takeda) Herausforderungen und praktische Lösungen im Zusammenhang mit aseptischen Prozesssimulationen (APS). Anna Campanella (Takeda) und Hussein Bachir (Franz Ziel) untersuchten, wie der Schutz von First Air in der aseptischen Verarbeitung durch die Anlagenkonstruktion, Handschuhinterventionen und Bedieneraktivitäten beeinträchtigt werden kann.
Wenn Ihr aseptischer Prozess wie ein Pferd aussieht, sieht Ihr APS wie ein Einhorn aus.
Diese einprägsame Analogie verwendete Alberto Gonzales (Takeda) während seines Vortrags auf der PDA Good Aseptic Manufacturing Conference 2026 (11.-12.06.2026). Darin erörterte er Herausforderungen und praktische Lösungsansätze im Zusammenhang mit aseptischen Prozesssimulationen (APS).
Gemäß EU-GMP-Anhang 1 sollte eine APS den routinemäßigen aseptischen Herstellungsprozess so genau wie möglich nachbilden. In der Praxis weisen APS-Läufe jedoch aufgrund regulatorischer Anforderungen bewusst eingebaute Unterschiede auf. So werden beispielsweise sterile Nährmedien anstelle des Produkts verwendet, transparente Behältnisse anstelle von opaken oder braunen sowie die befüllten Einheiten anschließend inkubiert.
Diese Abweichungen vom Routineprozess können ihrerseits zusätzliche Kontaminationsrisiken mit sich bringen und letztlich zu einer fehlgeschlagenen APS führen.

Abbildung 1 | Bewusste Abweichungen der APS im Vergleich zum Routineprozess (Quelle: Alberto Gonzales, PDA Good Aseptic Manufacturing Conference 2026)
Berücksichtigen Sie die Invalidierung von APS!
Anhang 1 schreibt vor, dass auf eine fehlgeschlagene APS mindestens drei aufeinanderfolgende erfolgreiche Wiederholungsläufe folgen müssen. Das Dokument enthält jedoch keine Aussagen dazu, unter welchen Umständen eine APS als ungültig erklärt bzw. invalidiert werden kann.
Der PDA Technical Report No. 22 (Process Simulation for Aseptically Filled Products) behandelt dieses Thema und stellt fest, dass eine APS invalidiert werden kann, wenn sie durch Bedingungen beeinträchtigt wurde, die normalerweise nicht Teil des routinemäßigen aseptischen Prozesses sind. In solchen Fällen kann die invalidierte APS durch einen einzelnen Wiederholungslauf ersetzt werden, ohne dass automatisch drei aufeinanderfolgende erfolgreiche APS erforderlich sind.
Die Unterscheidung zwischen einer fehlgeschlagenen APS und einer invalidierten APS ist von erheblicher Bedeutung, da sie Auswirkungen auf die Ursachenanalyse, die Bewertung des aseptischen Prozesses sowie den Umfang der erforderlichen Wiederholungsläufe haben kann.
First Air ist keine Selbstverständlichkeit – sie muss geschützt werden.
Anna Campanella (Takeda) und Hussein Bachir (Franz Ziel) zeigten, wie der Schutz der First Air in der aseptischen Verarbeitung durch die Konstruktion der Anlagen, Handschuhinterventionen und Bedienertätigkeiten beeinträchtigt werden kann.
Eine wichtige Erkenntnis aus Anna Campanellas Vortrag zum Thema First Air: Isolator-Design und -Betrieb müssen zusammenwirken, um die First Air über kritischen Oberflächen aufrechtzuerhalten. Nicht sterile Komponenten sollten so positioniert werden, dass sie den Luftstrom nicht stören, während die Betriebsabläufe Störungen und Turbulenzen minimieren sollten.
Hussein Bachir erläuterte, wodurch sich akzeptable von inakzeptablen Luftströmungsturbulenzen unterscheiden. Lokale Turbulenzen, die den Schutzschild der First Air nicht durchdringen, können akzeptabel sein. Turbulenzen, die den Erstluftschutz stören, können jedoch potenziell Kontaminationsrisiken mit sich bringen und müssen vermieden werden. Auch Turbulenzen unterhalb des kritischen Schutzpunktes (Second Air) gelten als inakzeptabel, wenn sie über den kritischen Schutzpunkt hinausreichen und den Erstluftschutz stören.
Ein weiterer wichtiger Aspekt seines Vortrags betraf den Einsatz von CFD (Computa-tional Fluid Dynamics). Numerische Strömungssimulationen können während der Planung und Risikobewertung wertvolle wissenschaftliche Erkenntnisse liefern, müssen jedoch im Rahmen der Qualifizierung durch Rauchstudien bestätigt werden.
Das Verständnis des Luftströmungsverhaltens bleibt ein Eckpfeiler der Kontaminationskontrolle.
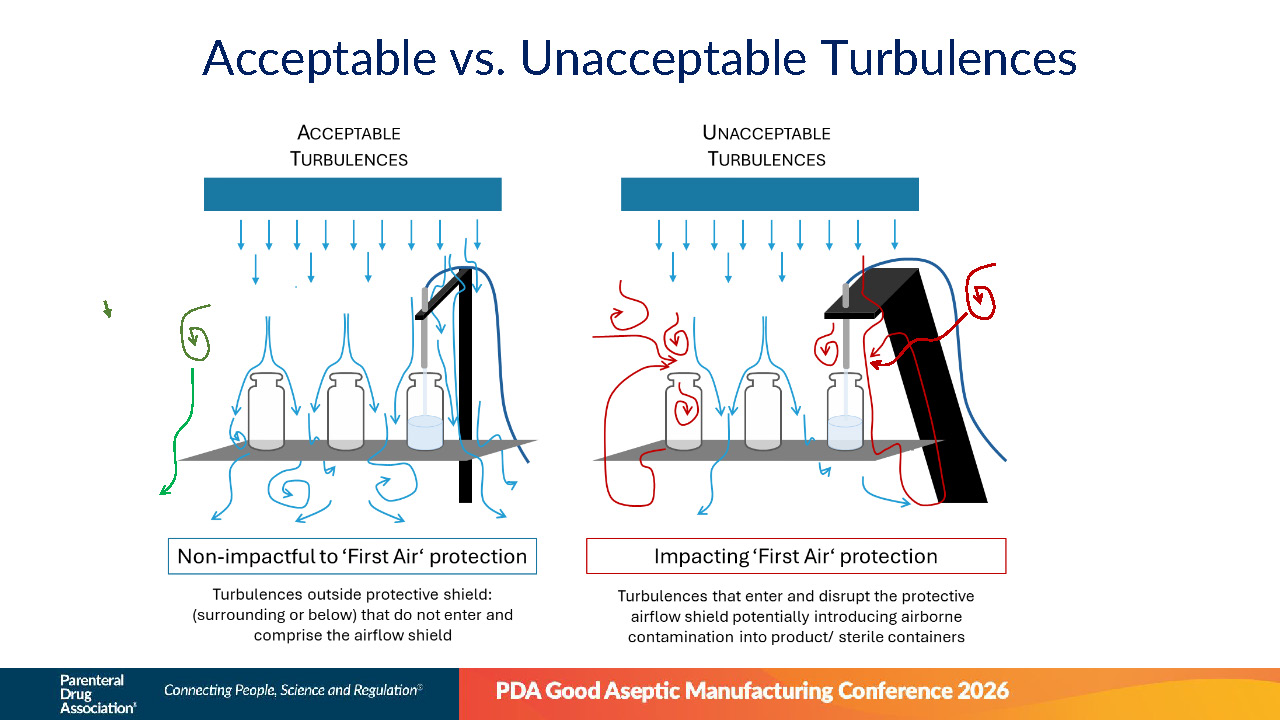
Abbildung 2 | Akzeptable versus inakzeptable Turbulenzen (Quelle: Hussein Bachir, PDA Good Aseptic Manufacturing Conference 2026)
Haben Sie Fragen oder Anregungen? Bitte kontaktieren Sie uns.redaktion@gmp-verlag.de








 zum EU-GMP-Leitfaden: Herstellung von sterilen Arzneimitteln")
")







